Gene Regulatory Network: Từ Phân Tử Đến Multi-Omics Và Ứng Dụng Lâm Sàng

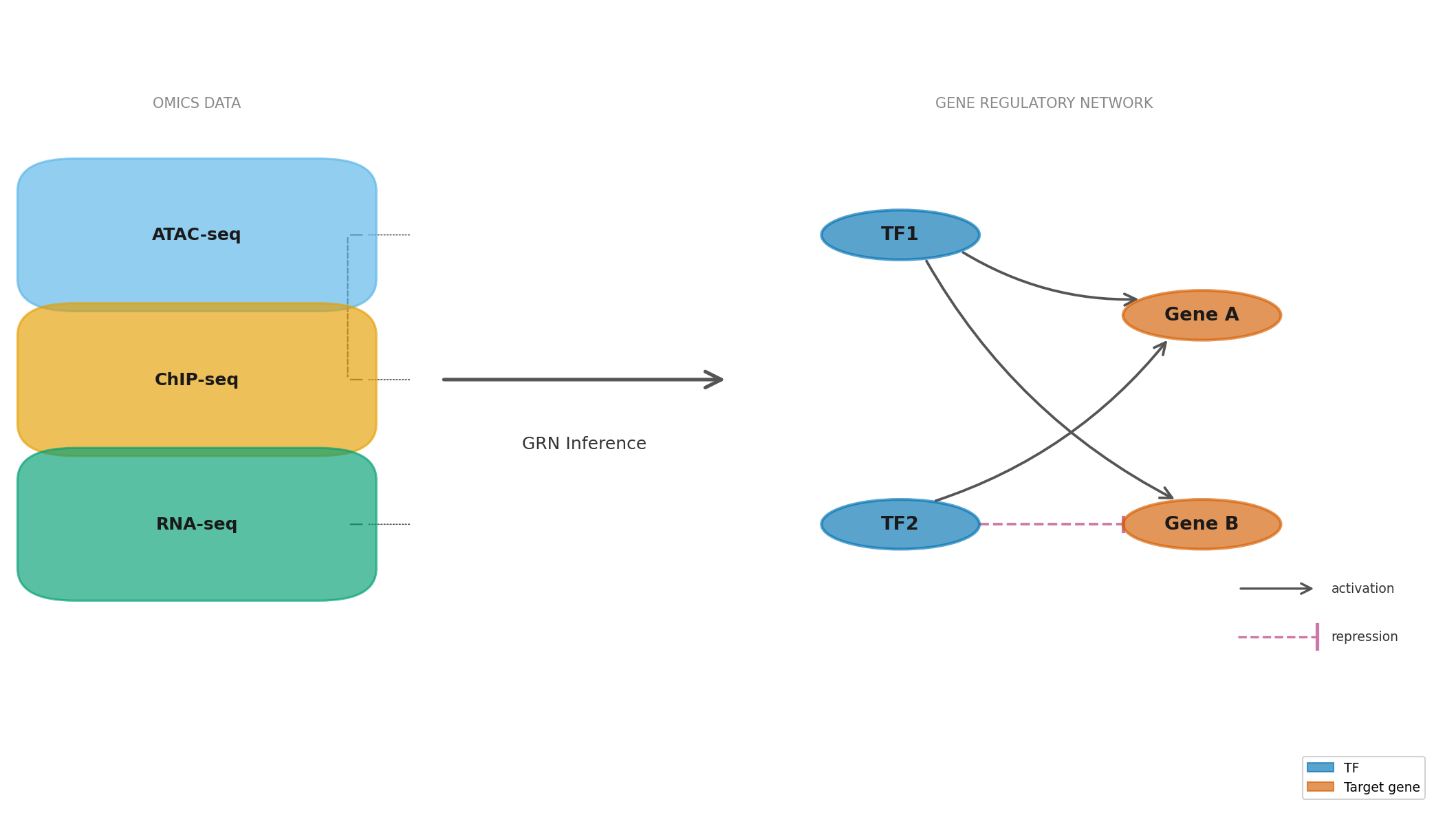
Genome của người chứa khoảng 20.000 gene mã hóa protein, nhưng không phải gene nào cũng hoạt động ở mọi tế bào trong mọi thời điểm. Một tế bào gan, một tế bào thần kinh, và một tế bào miễn dịch đều mang cùng một bộ DNA, nhưng chúng có hình dạng, chức năng, và tuổi thọ hoàn toàn khác nhau. Bí mật nằm ở gene regulatory network (GRN: mạng lưới điều hòa gene, hệ thống các phân tử và tín hiệu quy định việc gene nào được bật, gene nào bị tắt, và ở mức độ nào trong từng loại tế bào tại từng thời điểm cụ thể). GRN là lớp điều khiển cao nhất của sinh học tế bào — nó quyết định bản sắc tế bào, phản ứng với môi trường, và cả quá trình phát bệnh khi mạng lưới này bị rối loạn.
Hiểu GRN đòi hỏi không chỉ nắm khái niệm sinh học mà còn phải hiểu cơ sở toán học và thuật toán đằng sau từng công nghệ đo lường — bởi vì GRN không thể quan sát trực tiếp: nó phải được suy luận từ dữ liệu thực nghiệm thông qua các mô hình thống kê và học máy. Bài viết này dẫn dắt từ thành phần phân tử cơ bản nhất, qua phân tích thuật toán chi tiết của từng phương pháp omics, đến multi-omics integration và các ứng dụng thực tiễn vào năm 2026.
1. Khái Niệm Nền Tảng
1.1. Định Nghĩa Và Cấu Trúc Mạng Lưới
Một GRN về bản chất là một đồ thị có hướng có trọng số (weighted directed graph) trong đó mỗi node đại diện cho một gene (hay sản phẩm của nó), và mỗi edge mang hướng, dấu, và trọng số. Nếu transcription factor (TF: nhân tố phiên mã, là protein gắn vào DNA để bật hoặc tắt quá trình sản xuất RNA từ một gene) A kích hoạt biểu hiện gene B với cường độ w, ta biểu diễn cạnh A → B với trọng số +w. Nếu TF C ức chế gene D, cạnh C → D mang trọng số âm.
Biểu diễn toán học đơn giản nhất là ma trận kề có dấu (signed adjacency matrix) A kích thước N × N, trong đó A[i,j] là cường độ điều hòa của gene i lên gene j. Ma trận này trong thực tế là cực thưa (sparse): mỗi TF chỉ trực tiếp điều hòa hàng chục đến vài trăm gene, trong khi genome chứa hàng chục nghìn gene. Tính thưa này là tính chất thiết yếu — nó phản ánh tính đặc hiệu của TF và giúp hệ thống đủ ổn định khi bị nhiễu.
Trong thực tế, GRN không phẳng mà nhiều tầng: các gene mã hóa TF, các TF đó lại điều hòa các TF khác, tạo nên hệ thống phân cấp. Các master regulator (TF chủ chốt) ở đỉnh hệ thống phân cấp điều phối hàng nghìn gene thứ cấp thông qua các TF trung gian. Phân cấp này tạo nên cái gọi là bow-tie architecture (kiến trúc nơ lông): một tầng nhỏ các master TF fanout ra kiểm soát số lượng lớn gene đầu ra, và sự phá vỡ ở tầng master gây hiệu ứng thác cascade toàn bộ mạng.
1.2. Tại Sao GRN Quan Trọng
Khi các nhà khoa học giải mã genome người năm 2003, điều bất ngờ nhất không phải là 20.000 gene protein-coding — mà là 98% genome còn lại không mã hóa protein. Phần lớn vùng DNA này, từng bị gọi là “junk DNA”, thực ra chứa hàng triệu cis-regulatory element (CRE: yếu tố điều hòa cis, vùng DNA không mã hóa nhưng điều phối việc bật/tắt gene lân cận bằng cách gắn kết các TF và co-activator). Dự án ENCODE đã xác định hơn 1,2 triệu CRE tiềm năng trong genome người — gấp ~60 lần số gene protein-coding. Điều này lật ngược quan điểm cũ: thay vì “gene là tất cả”, sinh học hiện đại nhận ra rằng cách gene được điều hòa quan trọng không kém cấu trúc của gene đó.
Bằng chứng lâm sàng rõ ràng nhất đến từ các nghiên cứu genome-wide association study (GWAS: nghiên cứu liên kết toàn genome tìm các biến thể di truyền liên quan đến bệnh lý). Hơn 80% các biến thể di truyền liên quan đến bệnh — kể cả ung thư, tiểu đường, bệnh tim — nằm trong các vùng không mã hóa protein. Chúng không phá vỡ cấu trúc protein, nhưng chúng phá vỡ CRE, tức là phá vỡ một nút trong GRN. Đây là lý do tại sao giải mã GRN trở thành mục tiêu trung tâm của y học phân tử hiện đại.
2. Các Thành Phần Phân Tử Của GRN
2.1. Transcription Factors Và Cơ Chế Gắn DNA
TF là trung tâm của mọi GRN. Protein này chứa một DNA-binding domain (DBD: vùng gắn DNA, nhận diện một trình tự DNA ngắn đặc trưng gọi là motif, thường dài 6–12 nucleotide) và một activation/repression domain tương tác với bộ máy phiên mã hoặc co-repressor. Genome người mã hóa khoảng 1.639 TF (Lambert và cộng sự, 2018), mỗi TF nhận diện một tập hợp CRE khác nhau trên genome.
Để mô tả đặc tính gắn DNA của một TF, người ta dùng position weight matrix (PWM: ma trận trọng số vị trí). Đây là ma trận 4 × L trong đó L là chiều dài motif, hàng tương ứng với 4 nucleotide (A, C, G, T), và mỗi ô ghi log-odds score tại vị trí đó. Khi một TF “quét” DNA, nó tính tổng điểm PWM tại mỗi cửa sổ L nucleotide — chuỗi nào có tổng điểm vượt ngưỡng thì khả năng được gắn. Cơ sở vật lý đằng sau là thermodynamic binding: năng lượng gắn của TF lên một trình tự tỉ lệ thuận với tổng điểm PWM theo lý thuyết thống kê nhiệt động học. Điểm PWM cao đồng nghĩa với đóng góp năng lượng tự do âm (ΔG < 0), tức gắn ưu tiên về mặt nhiệt động học.
Điều làm cho TF thú vị về mặt mạng lưới là tính đặc hiệu theo ngữ cảnh (context-specificity). Cùng một TF GATA1 có thể kích hoạt bộ gene này trong tế bào hồng cầu nhưng kích hoạt bộ gene hoàn toàn khác trong tế bào megakaryocyte. Sự khác biệt đến từ các co-factor (đối tác cùng gắn DNA), trạng thái chromatin tại vùng đó (đóng hay mở), và cấu trúc không gian 3D genome. Một TF không hoạt động đơn lẻ mà luôn hoạt động trong combinatorial logic — sự kết hợp của 2–5 TF tại một CRE mới tạo nên quyết định cuối cùng về việc bật hay tắt gene.
2.2. Cis-Regulatory Elements
Promoter (vùng khởi động: vùng DNA ngay phía trên điểm bắt đầu phiên mã, nơi RNA polymerase II và các TF cơ bản — TFIID, TFIIB, TFIIH — lắp ráp thành pre-initiation complex để bắt đầu tổng hợp RNA) là CRE quen thuộc nhất. Cấu trúc promoter điển hình bao gồm TATA box tại vị trí −31/−26 (nếu có), Initiator element xung quanh điểm bắt đầu phiên mã (+1), và downstream promoter element (DPE) tại +28/+32. Không phải gene nào cũng có TATA box — khoảng 30% gene người có promoter kiểu TATA-less được điều hòa bởi các CpG island.
Nhưng promoter chỉ là một loại CRE trong hệ sinh thái điều hòa phong phú hơn nhiều:
- Enhancer (tăng cường tử: vùng DNA xa gene mục tiêu — đến vài trăm kilobase — nhưng tiếp xúc vật lý với promoter qua vòng chromatin để đưa TF-activator đến gần bộ máy phiên mã). Một gene có thể được điều tiết bởi hàng chục enhancer hoạt động trong các loại tế bào hay điều kiện khác nhau. Paradigm gần đây cho thấy enhancer không chỉ “mở khóa” gene mà còn đóng vai trò liquid–liquid phase separation (phân tách pha lỏng–lỏng: các TF co-activator BRD4, Mediator, và RNA polymerase II đồng ngưng tụ thành giọt lỏng nhỏ tại super-enhancer, từ đó nồng độ cục bộ cao giúp phiên mã hiệu quả hơn).
- Silencer (trình tự im lặng: CRE khi gắn bởi repressor TF — điển hình là phức hợp PRC2 polycomb — sẽ đặt histone mark H3K27me3 ức chế phiên mã).
- Insulator (trình tự cách ly: CRE ngăn tín hiệu kích hoạt từ enhancer “tràn” sang gene không phải mục tiêu). Protein CTCF gắn vào insulator và tương tác với cohesin để hình thành CTCF-mediated chromatin loop — ranh giới chức năng trong không gian 3D. Đột biến CTCF binding site có thể xóa ranh giới TAD, khiến enhancer của một gene “xuyên biên” kích hoạt oncogene ở vùng lân cận — cơ chế này phổ biến trong ung thư máu và não.
2.3. Kiến Trúc Không Gian Ba Chiều
GRN không thể hiểu trọn vẹn nếu chỉ xem genome như một chuỗi 1D. Trong nhân tế bào, DNA 2 mét được nén vào nhân đường kính 6–10 µm nhờ gấp cuộn theo thứ bậc không ngẫu nhiên:
Mức nucleosome: DNA quấn quanh octamer histone (H2A, H2B, H3, H4). Trạng thái acetylation của đuôi histone H3K27ac và H3K4me1 đánh dấu enhancer đang hoạt động; H3K4me3 đánh dấu promoter; H3K27me3 và H3K9me3 đánh dấu chromatin đóng. Đây là lớp thông tin epigenomics — không thay đổi trình tự DNA nhưng thay đổi cách genome được đọc.
Topologically associating domain (TAD: miền liên kết theo cấu trúc, vùng genome dài 0,1–1 megabase trong đó các đoạn DNA tương tác với nhau nhiều hơn với DNA bên ngoài miền). TAD là đơn vị cấu trúc cơ bản của genome: một enhancer trong TAD của gene A sẽ ưu tiên tương tác với gene A, ít tương tác với gene B ở TAD khác. Ranh giới TAD được neo bởi các vị trí gắn CTCF hội tụ (convergent CTCF binding: hai site hướng về nhau tạo neo cấu trúc cho cohesin loop extrusion).
A/B compartment (phân khu A/B: ở mức độ megabase, genome phân thành vùng A — chromatin mở, giàu gene, phiên mã tích cực — và vùng B — chromatin đóng, gene câm lặng, thường gần lamina nhân). Khi tế bào biệt hóa, nhiều vùng B chuyển sang A hoặc ngược lại, kéo theo thay đổi biểu hiện hàng trăm gene.
2.4. Motif Mạng Lưới Và Toán Học Ổn Định
Không phải mọi cấu trúc cục bộ trong GRN đều xuất hiện ngẫu nhiên. Một số network motif (môtif mạng: mẫu kết nối lặp lại trong mạng nhiều hơn kỳ vọng ngẫu nhiên) có chức năng tính toán đặc trưng. Quan trọng nhất là toggle switch — hai TF ức chế lẫn nhau — giải thích toán học về quyết định cell fate.
Hệ phương trình vi phân thường (ODE) mô tả toggle switch giữa hai TF x₁ và x₂:
\[\frac{dx_1}{dt} = \frac{\alpha_1}{1 + x_2^n} - \delta_1 x_1\] \[\frac{dx_2}{dt} = \frac{\alpha_2}{1 + x_1^n} - \delta_2 x_2\]Trong đó α là tốc độ sản xuất ở trạng thái không bị ức chế, δ là tốc độ phân hủy, và n là Hill coefficient (hệ số Hill, đo mức độ cooperativity của sự ức chế). Khi n > 1, hàm Hill ức chế trở nên cạnh hơn ở ngưỡng, phản ánh thực tế rằng nhiều phân tử repressor cần gắn cùng lúc để tắt hoàn toàn gene.
Phân tích điểm cố định cho thấy khi n đủ lớn, hệ có hai điểm cố định ổn định (x₁ cao / x₂ thấp, và x₁ thấp / x₂ cao) cùng một điểm cố định không ổn định ở giữa — đây là bistability. Tế bào “nhảy” giữa hai trạng thái khi nhận tín hiệu đủ mạnh, và một khi đã chọn trạng thái, nó duy trì ổn định ngay cả khi tín hiệu ban đầu mất đi — cơ chế của cell memory và quyết định biệt hóa.
Feedforward loop (FFL) là motif phổ biến thứ hai: TF A điều hòa cả B và C, đồng thời B cũng điều hòa C. Loại coherent type-1 FFL (A và B đều kích hoạt C) lọc nhiễu ngắn: chỉ khi tín hiệu A duy trì đủ lâu để tăng B thì C mới bật, ngăn phản ứng với xung tín hiệu thoáng qua. Ngược lại, incoherent type-1 FFL (A kích hoạt B và C, nhưng B ức chế C) tạo pulse biểu hiện C ngắn: C tăng nhanh khi A xuất hiện, rồi giảm dần khi B tích lũy đủ — hữu ích cho các gene cần kích hoạt tức thời nhưng không cần duy trì.
| Motif | Cấu trúc logic | Chức năng |
|---|---|---|
| Toggle switch | A ⊣ B, B ⊣ A | Cell fate decision, bistability |
| Autoregulation âm | A ⊣ A | Ổn định biểu hiện, giảm noise |
| Autoregulation dương | A → A | Khuếch đại tín hiệu, ổn định trạng thái |
| Coherent FFL type 1 | A→B, A→C, B→C | Lọc nhiễu tín hiệu ngắn |
| Incoherent FFL type 1 | A→B, A→C, B⊣C | Tạo pulse biểu hiện tức thời |
Bảng 2.4. Các motif mạng lưới phổ biến và cơ chế tính toán của chúng.
3. Đo Lường GRN: Công Nghệ Và Thuật Toán Chi Tiết
3.1. ChIP-seq Và Thuật Toán Gọi Peak MACS2
ChIP-seq (Chromatin Immunoprecipitation sequencing: kết hợp kết tủa miễn dịch chromatin với giải trình tự thế hệ mới để tạo bản đồ genome-wide về vị trí gắn kết của một TF hoặc histone mark cụ thể) là tiêu chuẩn vàng để lập bản đồ TF–DNA binding. Nguyên lý: cố định protein–DNA bằng formaldehyde, phân mảnh chromatin, rồi dùng kháng thể đặc hiệu kéo xuống phức hợp protein–DNA, giải trình tự DNA phần bị kéo xuống, và định vị các đỉnh (peaks) để biết TF đó gắn ở đâu trên genome.
Kết quả là hàng triệu đoạn đọc ngắn (reads) chỉ định vị trí trên genome — nhưng việc tìm peak (vùng làm giàu thực sự chứ không phải nhiễu nền) đòi hỏi thuật toán thống kê nghiêm ngặt. MACS2 là thuật toán gọi peak phổ biến nhất; quá trình hoạt động cụ thể từng bước như sau:
Bước 1: Xây dựng pileup signal. MACS2 đọc các read ChIP-seq và “dịch” vị trí về phía 5’ để tái tạo tâm của fragment gốc (~200 bp). Sau đó đếm số read trùng lên tại từng vị trí genome để tạo signal track 1D.
Bước 2: Ước tính background cục bộ (local lambda). Để phân biệt peak thật với vùng có nhiều read do bias ngẫu nhiên, MACS2 ước tính tốc độ background λ theo 4 mức: toàn genome (λ_BG), cửa sổ 100 kb (λ_100k), 10 kb (λ_10k), và 1 kb (λ_1k). Lambda thực sự dùng để gọi peak là λ_local = max(λ_BG, λ_10k, λ_1k) — lấy giá trị lớn nhất để đảm bảo conservative, tránh false positive ở những vùng genome vốn đã có read density cao do hiệu ứng vật lý.
Bước 3: Kiểm định thống kê Poisson. Với giả định reads phân phối Poisson với tham số λ_local, MACS2 tính p-value cho mỗi cửa sổ 200 bp có signal cao hơn background:
\[p = P(X \geq k \mid \lambda_{local}) = 1 - \sum_{i=0}^{k-1} \frac{e^{-\lambda} \lambda^i}{i!}\]trong đó k là số read quan sát trong cửa sổ đó.
Bước 4: Kiểm soát FDR toàn genome. Vì số lượng kiểm định song song cực lớn (~3 triệu cửa sổ trong genome người), MACS2 áp dụng Benjamini-Hochberg correction để chuyển đổi p-value sang q-value (false discovery rate). Các vùng có q-value < 0,05 được khai báo là peak. Summit của peak (điểm có signal cao nhất) được lưu để downstream analysis.
Control/Input sample (mẫu đối chứng, thường là DNA từ cùng tế bào nhưng không dùng kháng thể) được dùng song song để ước lượng λ chính xác hơn, loại bỏ các vùng genome có khả năng bắt giữ cao do bias cấu trúc chromatin chứ không phải TF binding thực sự. Đây là điểm khác biệt quan trọng của MACS2 so với phương pháp threshold đơn giản.
3.2. ATAC-seq: Cơ Chế Tn5 Và Phân Tích Vùng Mở
ATAC-seq (Assay for Transposase-Accessible Chromatin with sequencing: sử dụng enzyme transposase Tn5 để cắt và gắn adapter vào những vùng chromatin hở — tức vùng không bị nucleosome che phủ — sau đó giải trình tự để lập bản đồ chromatin accessibility toàn genome) là công nghệ đột phá vì chỉ cần ~50.000 tế bào (so với hàng triệu cho DNase-seq trước đây).
Cơ chế Tn5 tagmentation: Tn5 transposase là enzyme vi khuẩn được nạp sẵn hai adapter sequencing. Nó xâm nhập vào vùng chromatin mở rồi cắt và gắn adapter tại cùng một lúc (tagmentation). Vùng DNA bị bao phủ bởi nucleosome không thể bị Tn5 cắt vì enzyme không tiếp cận được. Kết quả: chỉ các vùng nucleosome-free mới được đọc.
Fragment size distribution sau ATAC-seq cực kỳ đặc trưng và chứa thông tin về cấu trúc nucleosome:
- < 100 bp: fragment từ vùng nucleosome-free region (NFR) — CRE đang hoạt động, TF binding site thực sự
- ~180 bp: fragment bao phủ một nucleosome (mono-nucleosomal)
- ~360 bp: fragment bao phủ hai nucleosome (di-nucleosomal)
- ~540 bp: ba nucleosome (tri-nucleosomal)
Pattern ladder này xuất hiện vì vùng chromatin giữa hai nucleosome (linker DNA) cũng bị Tn5 cắt, nhưng chỉ các fragment NFR mới là markers mạnh cho vùng mở thực sự. Phân tích chỉ dùng NFR-sized fragments cho độ phân giải peak cao hơn đáng kể.
TF footprinting là kỹ thuật tinh tế hơn: một TF đang thực sự gắn DNA sẽ bảo vệ ~15–20 bp dưới footprint của nó, tạo “dip” nhỏ trong signal ATAC-seq ngay tại motif, được flanked bởi signal cao hơn (do Tn5 cắt nhiều ngay cạnh vùng bị bảo vệ). Công cụ TOBIAS quét mọi motif TF đã biết trên toàn genome và nhận diện pattern dip/flank này để dự đoán TF nào đang thực sự gắn tại từng vị trí, phân biệt với các TF chỉ có motif nhưng không gắn trong điều kiện đó. Tn5 insertion bias (enzyme không cắt hoàn toàn ngẫu nhiên về trình tự) phải được hiệu chỉnh trước khi footprinting để tránh false signal.
3.3. Hi-C Và Thuật Toán Giải Mã Cấu Trúc 3D Genome
Hi-C (High-throughput Chromosome Conformation Capture: kỹ thuật đo lường toàn genome về tần số tương tác giữa các cặp loci trong nhân tế bào) tạo ra contact matrix C kích thước N × N trong đó C[i,j] là số read ghi nhận tương tác giữa bin i và bin j, với mỗi bin thường là 5 kb–50 kb.
Chuẩn hóa KR (Knight-Ruiz): Contact matrix thô có nhiều bias hệ thống — vùng GC cao, độ dài restriction fragment, khả năng bắt giữ của probe. Thuật toán KR tìm vector bias factor b sao cho ma trận chuẩn hóa O[i,j] = C[i,j] / (b[i] × b[j]) thỏa mãn tổng hàng và cột bằng nhau (doubly stochastic matrix). Bước này loại bỏ bias kỹ thuật không cần biết nguyên nhân cụ thể.
Gọi TAD bằng insulation score: Tại mỗi vị trí i, tính tổng contact giữa tất cả loci trong cửa sổ [i−W, i] và [i, i+W]. Vị trí có insulation score thấp (cửa sổ ít contact xuyên qua đó) là biên giới TAD. Minimum cục bộ trên đường cong insulation score cho vị trí ranh giới TAD tự động. Các biên giới này thường trùng với convergent CTCF binding sites (hai site CTCF hướng về nhau) và mật độ cao H3K4me1/CTCF peaks trong ChIP-seq.
Phát hiện A/B compartment bằng phân tích eigenvector: Hi-C matrix sau khi chuẩn hóa được chuyển sang correlation matrix (Pearson correlation giữa mọi cặp cột/hàng), rồi phân tích eigenvector đầu tiên (PC1). Dấu của PC1 phân chia genome thành compartment A (PC1 > 0, chromatin mở) và B (PC1 < 0, chromatin đóng). Dấu được xác nhận bằng cách so sánh với H3K27ac ChIP-seq hay mật độ gene — vùng giàu gene hơn được gán dấu A.
Loop calling với HiCCUPS: Các enhancer-promoter loop (tương tác điểm-điểm cụ thể) được gọi bằng HiCCUPS: tìm điểm trong contact matrix có giá trị cao hơn đáng kể so với vùng lân cận theo 4 hướng (donut background model), dùng kiểm định Poisson. HiChIP (kết hợp ChIP của H3K27ac với Hi-C) cho phép phát hiện enhancer-promoter loop hiệu quả hơn nhiều do làm giàu signal tại active enhancer trước khi đo contact.
3.4. Suy Luận GRN Từ RNA-seq: ARACNE Và GENIE3
RNA-seq đo biểu hiện gene nhưng không trực tiếp cho biết gene nào điều hòa gene nào. Suy luận GRN từ RNA-seq dựa trên giả định: nếu TF A điều hòa gene B, biểu hiện của A và B sẽ có mối quan hệ thống kê nhất định qua nhiều mẫu.
ARACNE dùng mutual information (thông tin hỗ tương: đo mức độ phụ thuộc thống kê giữa hai biến, bao gồm cả quan hệ phi tuyến, không giả định phân phối bình thường):
\[I(X; Y) = \sum_{x,y} p(x, y) \log \frac{p(x, y)}{p(x)p(y)}\]ARACNE xây dựng đồ thị đầy đủ giữa tất cả TF và gene, mỗi cạnh có trọng số MI[TF, gene]. Vấn đề với đồ thị đầy đủ là quá nhiều indirect interaction: nếu A điều hòa B và B điều hòa C, ta cũng quan sát MI(A;C) > 0 dù không có cạnh trực tiếp A→C.
ARACNE giải quyết bằng Data Processing Inequality (DPI): với bộ ba (A, B, C), nếu I(A;C) ≤ min(I(A;B), I(B;C)), cạnh A–C bị coi là gián tiếp và bị xóa. Lý do toán học: trong chuỗi Markov A → B → C, thông tin không thể tăng thêm khi truyền qua trung gian B. Áp dụng DPI lặp lại trên toàn bộ bộ ba loại bỏ phần lớn cạnh gián tiếp, cho GRN thưa và trực tiếp hơn.
GENIE3 tiếp cận như bài toán regression: với mỗi gene mục tiêu g, train một mô hình Random Forest để dự đoán biểu hiện của g từ biểu hiện của tất cả TF. Feature importance từ Random Forest đo đóng góp của mỗi TF vào khả năng dự đoán:
\[\text{Importance}(TF_t \to g) = \frac{1}{T} \sum_{k=1}^{T} \Delta_{\text{impurity}}(TF_t, \text{tree}_k)\]trong đó T là số cây, và Δ_impurity là mức giảm variance khi dùng TF_t để split. TF có importance cao được coi là “điều hòa” g mạnh hơn. Random Forest nắm bắt được tương tác phi tuyến và không nhạy với multicollinearity — điểm yếu của regression tuyến tính khi nhiều TF tương quan nhau. GRNBoost2 (cải tiến từ GENIE3 dùng gradient boosting trees) nhanh hơn nhiều khi xử lý single-cell data hàng nghìn tế bào.
4. Single-Cell Omics Và Suy Luận GRN
4.1. Tại Sao Cần Single-Cell
Phần lớn dữ liệu omics cổ điển là bulk data — trung bình hóa trên hàng triệu tế bào cùng lúc. Điều này che giấu tính dị chất tế bào (cellular heterogeneity): trong một khối u hay một mô phức tạp, hàng chục loại tế bào khác nhau tồn tại cùng nhau, mỗi loại có GRN riêng biệt. Bulk RNA-seq cho ra tín hiệu hỗn hợp không thể phân giải.
scRNA-seq với công nghệ droplet-based (điển hình là 10x Genomics Chromium) giải quyết vấn đề này. Nguyên lý: tế bào được đưa vào vi kênh với tốc độ thấp sao cho mỗi giọt dầu (droplet nước-trong-dầu) chỉ chứa tối đa một tế bào và một hạt micro-bead mang barcode DNA độc nhất. Khi tế bào vỡ trong droplet, mRNA của tế bào gắn vào barcode trên bead → sau đó giải trình tự toàn bộ bead, mỗi trình tự mang barcode tế bào + unique molecular identifier (UMI: trình tự ngẫu nhiên ngắn định danh từng phân tử RNA riêng lẻ để loại bỏ PCR duplicate bias). Kết quả là ma trận gene × tế bào với hàng chục nghìn tế bào — nền tảng để xây dựng GRN riêng cho từng loại tế bào.
4.2. scATAC-seq Và Thách Thức Dữ Liệu Thưa
scATAC-seq lập bản đồ chromatin accessibility riêng cho từng tế bào. Thách thức kỹ thuật lớn nhất là sparsity (tính thưa): genome người có ~6 triệu vị trí Tn5 insertion site tiềm năng, nhưng một tế bào đơn lẻ chỉ có ~2 bản sao mỗi locus, và chỉ một tỉ lệ nhỏ loci mở trong bất kỳ tế bào nào — dẫn đến ma trận peak × tế bào có đến 97–99% giá trị bằng 0.
Để xử lý sparsity, LSA/TF-IDF (Latent Semantic Analysis / Term Frequency-Inverse Document Frequency: phương pháp giảm chiều từ văn bản, được chuyển sang genomics vì ma trận peak × tế bào tương tự ma trận từ × tài liệu) chuẩn hóa và giảm chiều dữ liệu scATAC-seq. TF-IDF chuẩn hóa mỗi peak dựa trên tần suất xuất hiện trong từng tế bào và độ phổ biến trên toàn bộ dataset; sau đó SVD (singular value decomposition) giảm xuống 20–50 chiều tiềm ẩn phản ánh chương trình chromatin. LDA (Latent Dirichlet Allocation) cũng được dùng để phát hiện “topic” chromatin — mỗi topic là một chương trình mở chromatin đặc trưng cho một nhóm tế bào.
4.3. Multiome: Đo Đồng Thời RNA Và Chromatin
10x Genomics Multiome ATAC + Gene Expression đo đồng thời scRNA-seq và scATAC-seq từ cùng một tế bào bằng cách phân tách nhân tế bào trước khi tagmentation (nhân đi vào ATAC protocol) và giải phóng mRNA (đi vào RNA protocol) từ cùng một tế bào. Điều này loại bỏ vấn đề batch effect khi kết hợp hai thí nghiệm riêng lẻ và cho phép phân tích nhân quả trực tiếp hơn: biết chromatin accessibility của tế bào này → biết gene nào đang biểu hiện từ cùng tế bào đó. Năm 2025–2026, multiome đã trở thành tiêu chuẩn cho các nghiên cứu cell atlas quy mô lớn.
4.4. SCENIC: Pipeline Ba Bước Từ Biểu Hiện Đến Regulon
SCENIC (Single-Cell rEgulatory Network Inference and Clustering) là pipeline ba bước để xây dựng và ước tính hoạt động của TF regulon từ dữ liệu scRNA-seq, kết hợp thông tin biểu hiện với cơ sở dữ liệu motif.
Bước 1 — GRN inference. SCENIC dùng GRNBoost2 (hoặc GENIE3) để tính trọng số điều hòa giữa từng cặp (TF, target gene) từ ma trận biểu hiện. Kết quả là một danh sách TF cùng các “module” gene đồng biểu hiện với TF đó — nhưng đây mới chỉ là tương quan, chưa phải mạng nhân quả.
Bước 2 — Điều chỉnh bằng motif enrichment (RcisTarget). Để loại bỏ tương quan giả, SCENIC kiểm tra xem vùng regulatory (các ATAC peak hoặc ±10 kb từ TSS) của từng bộ target gene có enriched motif của TF tương ứng không. RcisTarget tính normalized enrichment score (NES) bằng cách xếp hạng tất cả gene theo điểm motif trong vùng của chúng, rồi tính diện tích dưới đường cong AUC cho gene trong module. Chỉ các module có NES ≥ 3 (hoặc ngưỡng tương đương) mới được giữ lại và gọi là regulon — tập hợp (TF, target genes) với bằng chứng cả tương quan biểu hiện lẫn motif TF trong vùng regulatory của target.
Bước 3 — Tính điểm hoạt động regulon per-cell (AUCell). AUCel tính Area Under the recovery Curve (AUC) cho mỗi regulon trong từng tế bào: các gene trong tế bào được xếp hạng theo biểu hiện (từ cao đến thấp), sau đó tính tỉ lệ gene thuộc regulon trong top-k gene được biểu hiện cao nhất. AUC cao ↔ regulon đang hoạt động trong tế bào đó. Kết quả là ma trận regulon activity (regulon × tế bào) có thể dùng để phân loại tế bào và xác định TF master regulator đặc trưng cho từng cluster.
4.5. CellOracle: GRN Hồi Quy Và Mô Phỏng Perturbation
CellOracle (Kamimoto và cộng sự, 2023) xây dựng GRN bằng cách kết hợp scATAC-seq để xác định cấu trúc mạng cơ sở, sau đó dùng scRNA-seq để ước tính trọng số kết nối.
Bước 1 — Base GRN từ scATAC-seq. Với mỗi peak ATAC, CellOracle quét motif TF (từ JASPAR) và xác định TF nào có motif trong peak đó. Sau đó, peak được liên kết với gene target gần nhất (hoặc dùng dữ liệu Hi-C để chỉ định target đúng hơn). Kết quả: cạnh TF → gene cho những cặp có cả bằng chứng motif lẫn chromatin accessibility.
Bước 2 — Học trọng số bằng ridge regression. Với mỗi gene g, CellOracle fit mô hình tuyến tính hồi quy có penalization:
\[\hat{g} = \sum_{t \in \text{TF regulators of } g} \beta_t \cdot x_t\]trong đó x_t là biểu hiện của TF t và β_t là trọng số cần ước tính. Ridge regression (L2 regularization: thêm hạng phạt λ‖β‖² vào loss function để tránh overfitting khi số TF regulators nhiều hơn số mẫu) cho kết quả β_t phản ánh mức độ điều hòa thực tế. Ma trận toàn bộ β tạo thành ma trận kề GRN có trọng số.
Bước 3 — In silico perturbation. Để mô phỏng knock out TF k: đặt x_k = 0 rồi lan truyền hiệu ứng qua ma trận kề bằng phép nhân ma trận lặp (hoặc giải hệ tuyến tính). Sự thay đổi biểu hiện gene ∆g của mọi gene được tính và so sánh với dữ liệu thực nghiệm. Nếu vector ∆g dự đoán trùng khớp với vector quan sát trong Perturb-seq → mô hình GRN được xác nhận. Quan trọng hơn, in silico perturbation CellOracle cho phép dự đoán hướng biệt hóa tế bào gốc khi một TF bị kích hoạt hay bất hoạt, hỗ trợ thiết kế thí nghiệm reprogramming tối ưu.
5. Multi-Omics Integration Cho GRN
5.1. Điểm Mù Của Từng Omics Layer
Mỗi công nghệ omics nhìn vào GRN từ một góc độ riêng biệt và mỗi góc đều có điểm mù đặc trưng:
| Omics | Thông tin thu được | Điểm mù |
|---|---|---|
| ChIP-seq | TF gắn ở đâu | Binding có chức năng không? Kích hoạt hay ức chế? |
| ATAC-seq | CRE nào đang mở | TF nào đang sử dụng vùng mở đó? |
| RNA-seq | Gene nào biểu hiện | TF nào chịu trách nhiệm? Nhân quả hay tương quan? |
| Hi-C | Hai vùng DNA gần nhau trong 3D | Kết nối đó có chức năng điều hòa không? |
| proteomics | Protein nào hiện diện | Protein đó hoạt động như activator hay repressor? |
Bảng 5.1. Mỗi omics layer cung cấp thông tin bổ sung nhưng không đủ đứng một mình để xây dựng GRN đầy đủ.
Chỉ khi tích hợp đủ nhiều lớp, ta mới có thể suy luận GRN chính xác. Đây là lý do framework tích hợp multi-omics trở thành hướng nghiên cứu chủ đạo từ 2022 trở đi.
5.2. MOFA+: Mô Hình Nhân Tố Tiềm Ẩn Đa Omics
MOFA+ (Multi-Omics Factor Analysis Plus) là framework Bayesian học đồng thời từ K ma trận omics:
\[X_k \approx Z W_k^T + \epsilon_k, \quad k = 1, 2, \ldots, K\]trong đó X_k là ma trận omics thứ k (N mẫu × D_k features), Z là ma trận factor score (N × R, R là số nhân tố tiềm ẩn), W_k là ma trận loading của omics k (D_k × R), và ε_k là noise Gaussian. Mỗi factor (nhân tố tiềm ẩn) r đại diện cho một “chương trình sinh học” — ví dụ chu kỳ tế bào, chương trình stress, hay chương trình biệt hóa — được kích hoạt ở một tập con mẫu và phản ánh qua nhiều lớp omics đồng thời.
Điểm mấu chốt là sparse Bayesian priors trên W_k: prior spike-and-slab khuyến khích mỗi loading về 0 trừ khi có bằng chứng mạnh từ dữ liệu. Kết quả là mỗi factor chỉ được “giải thích” bởi một tập con features nhỏ — giúp diễn giải sinh học. MOFA+ mở rộng MOFA để xử lý nhiều nhóm mẫu (ví dụ bệnh nhân A và B) với các factor chia sẻ và riêng tư.
Từ góc độ GRN, các factor MOFA+ là “chữ ký multi-omics” của từng chương trình phiên mã: factor có loading cao ở RNA-seq và ATAC-seq của cùng một bộ enhancer-gene cặp tương ứng với một GRN module đang hoạt động.
5.3. LINGER: Deep Learning Từ Bulk Đến Single-Cell
LINGER (Larsson và cộng sự, 2024) xây dựng GRN qua hai giai đoạn. Giai đoạn 1: train mô hình deep learning trên dữ liệu bulk ENCODE quy mô lớn (hàng trăm loại tế bào với ChIP-seq, ATAC-seq, RNA-seq) để học mối quan hệ chung giữa chromatin accessibility và biểu hiện gene. Giai đoạn 2: fine-tune mô hình xuống dữ liệu single-cell multiome của mẫu cụ thể, tạo ra GRN có độ phân giải tế bào. Cách tiếp cận transfer learning này cho phép LINGER hoạt động tốt ngay cả với các loại tế bào ít dữ liệu, bởi vì mô hình đã học được các nguyên tắc điều hòa chung từ dữ liệu phong phú.
5.4. Spatial Multi-Omics Và GRN Trong Không Gian Mô
Năm 2025–2026, spatial transcriptomics kết hợp spatial ATAC-seq đã cho phép khám phá chiều mới: GRN thay đổi theo vị trí không gian của tế bào trong mô. Tế bào ở trung tâm khối u có GRN khác với tế bào ở rìa; tế bào viêm gần mạch máu có chromatin program khác với tế bào ở xa. MERFISH, Slide-tags, và Stereo-seq là các nền tảng spatial omics đang tạo ra spatial GRN atlas — bản đồ GRN kết hợp cả loại tế bào lẫn tọa độ không gian trong mô.
6. Deep Learning Và Foundation Models Cho GRN
6.1. Graph Neural Networks Và Message Passing
Graph neural network (GNN: mạng nơ-ron học trên dữ liệu đồ thị bằng cách lan truyền thông tin qua các cạnh kết nối để học biểu diễn của từng node trong ngữ cảnh láng giềng của nó) là kiến trúc phù hợp tự nhiên với GRN vì bản thân GRN đã là một đồ thị.
Message passing trong GNN hoạt động theo cơ chế: tại mỗi bước t, node i tổng hợp thông điệp từ tất cả node lân cận j ∈ N(i):
\[h_i^{(t+1)} = \text{UPDATE}\left(h_i^{(t)}, \text{AGGREGATE}\left(\{h_j^{(t)} : j \in \mathcal{N}(i)\}\right)\right)\]Sau nhiều bước lan truyền, embedding h_i^(T) của node i mã hóa thông tin về toàn bộ “vùng lân cận” trong mạng — cho phép mô hình học được rằng “TF A thường đồng điều hòa với TF B khi cả hai đều là downstream của master regulator C.” Các mô hình như DeepDRIM và GRNBoost2-GNN dùng GNN để suy luận cạnh mới trong GRN từ dữ liệu biểu hiện gene và cấu trúc network đã biết một phần.
6.2. Enformer: Sequence-To-Function Với Kiến Trúc CNN + Transformer
Enformer (Avsec và cộng sự, 2021) là mô hình dự đoán hoạt động của cis-regulatory element trực tiếp từ trình tự DNA, không cần đo thực nghiệm. Kiến trúc:
Input: 196.608 bp trình tự DNA dưới dạng one-hot encoding (4 × 196.608 tensor).
CNN trunk (dilated convolution): Nhiều lớp convolution 1D với dilation rate tăng dần (1, 2, 4, 8, 16…) để học pattern cục bộ (binding motif 6–20 bp) đồng thời dần dần mở rộng receptive field để nắm bắt context xa hơn. Mỗi lớp pooling giảm độ phân giải 2×, sau đó 4×, đến khi còn 896 bin × 768 channels.
Transformer trunk: 11 khối attention cho phép mọi cặp bin trong 896 × 896 tương tác với nhau — quan trọng để học rằng enhancer ở vị trí 80 kb có thể ảnh hưởng đến gene ở trung tâm. Multi-head self-attention với positional encoding cho phép nắm bắt những quan hệ đường dài mà CNN thuần túy không thể.
Output: Prediction của 5.313 genomic tracks tại 128 bp resolution — bao gồm RNA-seq, ATAC-seq, ChIP-seq của hàng trăm loại tế bào người và chuột. Tất cả được dự đoán đồng thời từ cùng một trình tự input.
Ứng dụng GRN: Bằng cách so sánh output khi thay thế từng nucleotide trong CRE (in silico mutagenesis), Enformer tính được contribution score của từng nucleotide đến biểu hiện gene — tức là “nucleotide nào trong enhancer quan trọng cho hoạt động của nó.” Điều này cho phép dự đoán tác động của biến thể di truyền (SNP) tại CRE lên biểu hiện gene, kết nối GWAS SNP với cơ chế GRN cụ thể.
Borzoi (2024) mở rộng Enformer với receptive field 524 kb và dự đoán RNA splicing, cho độ chính xác cao hơn đáng kể với các enhancer xa. AlphaGenome (2025) kết hợp kiến trúc AlphaFold-style với sequence-to-function prediction.
6.3. Foundation Models Cho Single-Cell
Geneformer (Theodoris và cộng sự, 2023) là mô hình BERT cho single-cell, được huấn luyện trên 29,9 triệu tế bào người. Cách tokenize đặc biệt: thay vì dùng giá trị biểu hiện thô (không thể so sánh qua batch do kỹ thuật khác nhau), Geneformer dùng rank-value tokenization — mỗi gene nhận token là thứ hạng biểu hiện của nó trong tế bào đó (gene biểu hiện cao nhất = token 1, gene tiếp theo = token 2, …). Cách này loại bỏ hoàn toàn batch effect và sequencing depth bias, vì chỉ thứ tự tương đối giữa các gene được giữ lại.
Pre-training: Geneformer dùng masked gene modeling (MGM) — tương tự masked language modeling trong BERT: ngẫu nhiên che ~15% gene trong sequence, yêu cầu mô hình dự đoán gene bị che từ context của các gene còn lại. Điều này buộc mô hình học được mạng điều hòa ngầm: để dự đoán gene bị che, mô hình phải suy ra gene nào thường co-expressed với nhau = học được GRN ẩn.
In silico perturbation với Geneformer: Để mô phỏng knock down TF k, Geneformer xóa token của k khỏi sequence và quan sát sự thay đổi trong embedding của các gene khác — gene nào thay đổi nhiều nhất khi mất TF k được coi là downstream target của k. Approach này đã xác định thành công drug target mới cho bệnh tim từ dữ liệu single-cell của bệnh nhân.
scGPT (2024) và scFoundation xây dựng trên paradigm tương tự với các cải tiến kiến trúc, và từ 2025 các mô hình này đang được tích hợp với dữ liệu lâm sàng để dự đoán drug response từ GRN của từng bệnh nhân.
6.4. Hạn Chế Của Các Mô Hình Deep Learning
Dù ấn tượng, các deep learning model cho GRN vẫn đối mặt với thách thức cơ bản. Interpretability (khả năng giải thích): mô hình dự đoán tốt nhưng không cho biết cơ chế đang hoạt động có giá trị hạn chế trong nghiên cứu cơ bản. Kỹ thuật attribution (SHAP, integrated gradients) giúp nhưng chưa đủ cụ thể. Distribution shift: nếu loại tế bào hay bệnh lý cần dự đoán khác xa phân phối huấn luyện, mô hình có thể “hallucinate” quan hệ không tồn tại. Scaling law chưa rõ ràng như trong NLP: tăng kích thước mô hình và dữ liệu chưa chắc cải thiện hiệu năng tương ứng. Kết hợp mechanistic ODE model với deep learning để lấy ưu điểm của cả hai đang là xu hướng nghiên cứu tích cực.
7. Mô Hình Toán Học Của GRN
7.1. Mô Hình ODE Và Phân Tích Điểm Cân Bằng
Ngoài các phương pháp thực nghiệm, GRN có thể được mô hình hóa toán học để dự đoán hành vi trong điều kiện chưa đo. Mô hình ODE (ordinary differential equation) là nền tảng: trạng thái của system tại thời điểm t được mô tả bởi vecto nồng độ x(t) = (x₁, x₂, …, xₙ) của n phân tử, với:
\[\frac{d\mathbf{x}}{dt} = \mathbf{f}(\mathbf{x})\]trong đó f(x) mô tả tốc độ thay đổi của từng thành phần dựa trên các tương tác điều hòa. Điểm cân bằng (steady state) x* thỏa f(x) = 0. Phân tích ổn định tuyến tính quanh x dựa trên ma trận Jacobian J = ∂f/∂x tại x: nếu tất cả eigenvalue của J có phần thực âm, x ổn định (hệ quay về sau khi bị nhiễu nhỏ); nếu có eigenvalue với phần thực dương, x* không ổn định.
Trong GRN, toggle switch có hai điểm ổn định tương ứng với hai trạng thái tế bào (xem Phần 2.4). Mô hình ODE dự đoán: ngưỡng tín hiệu cần thiết để chuyển đổi giữa hai trạng thái, thời gian chuyển đổi, và robustness (tỉ lệ có thể thay đổi parameter mà không làm mất bistability). Những dự đoán này có thể kiểm tra thực nghiệm bằng live-cell imaging theo dõi biểu hiện gene qua thời gian.
7.2. Boolean Networks Và Cảnh Quan Cell Fate
Boolean network là phiên bản đơn giản hóa: mỗi gene chỉ có hai trạng thái (ON = 1, OFF = 0) và được cập nhật theo logic rule dựa trên input từ các regulator. Ví dụ: gene A bật khi (TF1 = 1 AND TF2 = 1) OR (TF3 = 1 AND TF4 = 0). Mặc dù đơn giản hóa, Boolean networks nắm bắt được attractor — trạng thái ổn định của GRN tương ứng với cell type. Mỗi attractor là một vòng trạng thái (cycle) mà hệ hội tụ về sau nhiều bước cập nhật, bất kể xuất phát từ trạng thái ban đầu nào trong “basin of attraction” của nó.
Phân tích Boolean network của GRN phát triển phôi đã tái tạo thành công số lượng attractor khớp với số loại tế bào quan sát được — evidence đầy sức thuyết phục rằng GRN có đủ thông tin để “quyết định” tế bào trở thành loại tế bào nào mà không cần tín hiệu bên ngoài liên tục.
7.3. Mô Hình Stochastic Và Transcriptional Bursting
Trong thực tế tế bào đơn lẻ, phiên mã không phải là quá trình liên tục và trơn tru — nó diễn ra theo từng đợt tắt bật ngẫu nhiên, gọi là transcriptional bursting. Mô hình two-state promoter (promoter ngẫu nhiên chuyển qua lại giữa trạng thái active và inactive theo xác suất) mô tả điều này: khi promoter active, RNA được sản xuất với tốc độ k_on; khi inactive (tốc độ chuyển k_off), không có RNA nào được tạo ra.
Phân phối số lượng mRNA trong tế bào đơn lẻ là mixture distribution phản ánh cả hai trạng thái. Single-molecule FISH và scRNA-seq đo được phân phối này, cho phép ước tính k_on, k_off, và burst size (số RNA trung bình mỗi lần bật). Các TF activator thường tăng k_on (tần suất bật promoter) hoặc k_off^{-1} (kéo dài thời gian hoạt động), trong khi repressor làm ngược lại. Gillespie algorithm mô phỏng stochastic dynamics và cho phép dự đoán noise (biến động biểu hiện giữa các tế bào giống nhau) từ đặc tính GRN.
8. Ứng Dụng Lâm Sàng
8.1. GRN Trong Ung Thư Và Super-Enhancer Hijacking
Ung thư là bệnh lý GRN ở cấp độ cơ bản nhất. Một cơ chế quan trọng là super-enhancer hijacking (kẻ trộm super-enhancer): translocation chromosome hoặc đột biến điểm tại ranh giới TAD có thể đặt một proto-oncogene vào cùng TAD với một super-enhancer từ locus khác. Đột nhiên oncogene được kiểm soát bởi super-enhancer công suất cao — dẫn đến biểu hiện quá mức của oncogene. Trong ung thư T-cell acute leukemia, translocation đặt oncogene TAL1 dưới siêu cường tử của gene STILTAL1-SE, đây là cơ chế gốc của nhiều trường hợp bệnh.
Hiểu cơ chế này dẫn đến chiến lược điều trị: các chất ức chế BET bromodomain (BRD4 là thành phần của super-enhancer; BET inhibitor như JQ1 phá vỡ liquid-liquid condensate của super-enhancer, tắt đồng thời nhiều oncogene) đang trong thử nghiệm lâm sàng. Thách thức là tránh off-target vì BET inhibitor ảnh hưởng đến nhiều super-enhancer trên toàn genome.
Multi-omics integration trong ung thư cho phép xây dựng tumor-specific GRN: từ bulk RNA-seq + ChIP-seq + ATAC-seq + Hi-C của mẫu ung thư, xác định TF nào đang “lái” khối u, từ đó tìm thuốc nhắm vào TF hoặc CRE của nó. VIPER (Virtual Inference of Protein-activity by Enriched Regulon analysis) ước tính hoạt động của TF trong khối u từ RNA-seq bằng cách dùng regulon từ ARACNE — cho phép so sánh “TF activity landscape” giữa khối u và mô thường.
8.2. Drug Target Discovery Dựa Trên Network
Network medicine xem bệnh là rối loạn module trong mạng lưới sinh học, thay vì rối loạn một phân tử đơn lẻ. Để tìm drug target từ GRN:
Network centrality scoring: TF ở vị trí trung tâm cao trong GRN (betweenness centrality cao, in/out degree cao) điều phối nhiều gene bệnh hơn và là đòn bẩy hiệu quả hơn. Tuy nhiên, TF quá trung tâm thường thiết yếu cho tế bào bình thường, gây toxicity cao. Công cụ NetworkAnalyzer và Cytoscape visualize và tính centrality metrics tự động.
Synthetic lethality trong ngữ cảnh GRN: Một gene A được gọi là synthetic lethal với gene B nếu mất cả hai cùng lúc mới gây chết tế bào (mất một trong hai thì tế bào vẫn sống). Trong GRN, nếu hai TF điều hòa song song cùng một chương trình gene thiết yếu, tắt cả hai cùng lúc sẽ tắt hoàn toàn chương trình đó. Ung thư thường mất một trong hai đường — từ đó chỉ cần tắt đường còn lại là đủ giết tế bào ung thư mà không cần ảnh hưởng tế bào bình thường (còn đường back-up). CRISPR synthetic lethality screen kết hợp với GRN analysis hiện đangmở ra hướng drug target mới.
Direct reprogramming dùng in silico GRN (CellOracle, Geneformer) để thiết kế protocol chuyển đổi tế bào mà không cần qua giai đoạn tế bào gốc, giảm rủi ro khối u hóa. Protocol chuyển đổi fibroblast → cardiomyocyte đang ở giai đoạn thử nghiệm pre-clinical từ thiết kế in silico.
8.3. Epigenome Editing Và Can Thiệp Trực Tiếp Vào GRN
Từ 2023–2026, các công cụ biên tập epigenome đã đến mức đủ chính xác để sửa đổi trực tiếp GRN mà không cần thay đổi trình tự DNA:
- CRISPRa/CRISPRi: dCas9 không cắt DNA nhưng mang activator domain (VPR, SAM) hay repressor domain (KRAB) đến một CRE cụ thể, bật hay tắt gene mục tiêu reversibly.
- Base editing tại CRE: Adenine base editor hay cytosine base editor thay đổi nucleotide đơn lẻ trong motif TF tại CRE — đủ để phá vỡ hoặc tăng cường TF binding, điều chỉnh biểu hiện gene mà không cắt đôi DNA.
- Epigenome editing: dCas9 mang TET1 (demethylase) hay DNMT3A (methylase) đến promoter CpG island, thay đổi methylation và từ đó biểu hiện gene — dạng can thiệp GRN không để lại dấu vết trình tự.
9. Thách Thức Và Xu Hướng Năm 2026
9.1. Causality: Từ Tương Quan Đến Nhân Quả
Thách thức sâu nhất của GRN inference là hầu hết phương pháp hiện tại chỉ xác định tương quan (correlation), không phải nhân quả (causality). Perturb-seq (kết hợp CRISPR knock down với scRNA-seq để đo hệ quả biểu hiện gene sau khi knock down từng gene trong tập mục tiêu) là gold standard hiện nay để xác định nhân quả: nếu knock down TF A làm thay đổi biểu hiện gene B → A nhân quả điều hòa B.
Tuy nhiên, suy luận nhân quả từ dữ liệu quan sát (không can thiệp) có thể dùng các thuật toán học nhân quả PC algorithm (Peter-Clark algorithm: tìm DAG nhân quả bằng cách kiểm tra conditional independence giữa các biến và định hướng cạnh dựa trên v-structure) hoặc FCI (Fast Causal Inference: phiên bản mạnh hơn xử lý được hidden confounders). Các thuật toán này đang được áp dụng cho Perturb-seq data quy mô lớn để xây dựng causal GRN thay vì chỉ correlational GRN.
9.2. RNA Velocity Và GRN Theo Thời Gian
RNA velocity (La Manno và cộng sự, 2018) khai thác tỉ số unspliced RNA (pre-mRNA chưa được intron removed) và spliced mRNA để ước tính “chiều” và “tốc độ” thay đổi biểu hiện gene của mỗi tế bào. Vì unspliced RNA là RNA vừa mới được tổng hợp, so sánh nó với spliced RNA cho biết gene đang tăng hay giảm biểu hiện trong tương lai gần của tế bào đó.
\[\frac{du}{dt} = \alpha(t) - \beta u, \quad \frac{ds}{dt} = \beta u - \gamma s\]trong đó u là unspliced RNA, s là spliced mRNA, α là tốc độ phiên mã, β là tốc độ splicing, γ là tốc độ phân hủy. scVelo (Bergen và cộng sự, 2020) cải tiến RNA velocity bằng cách học các tham số α, β, γ động theo thời gian từ dữ liệu, thay vì giả định alpha không đổi.
CellRank kết hợp RNA velocity với Markov chain để dự đoán xác suất terminal state (trạng thái tế bào cuối cùng) của từng tế bào, và truy ngược lại để xác định initial progenitor state và các transition paths cell fate. Trong bối cảnh GRN, RNA velocity cho phép suy luận GRN theo chiều thời gian: TF nào thay đổi trước (initiators) và TF nào thay đổi sau (effectors) trong cascade biệt hóa.
9.3. Xu Hướng Nổi Bật Năm 2026
Perturbation atlas — tổng hợp kết quả Perturb-seq genome-wide qua nhiều loại tế bào thành cơ sở dữ liệu GRN nhân quả có độ phủ rộng. REmap 3.0 và ENCODE4 đang xây dựng atlas này với hàng trăm nghìn perturbation experiment. Mục tiêu: cứ mỗi gene trong genome, biết hệ quả biểu hiện của việc knock down nó ở mỗi loại tế bào chính.
End-to-end foundation models — kết hợp sequence-to-function model (Enformer/Borzoi) với single-cell foundation model (scGPT/Geneformer) để mô hình hóa GRN từ DNA sequence đến phenotype tế bào trong một framework duy nhất, không cần pipeline rời rạc. AlphaGenome (DeepMind, 2025) là bước đầu tiên theo hướng này.
Epigenetic aging và GRN rewiring — liên kết thay đổi GRN theo tuổi tác (tái lập trình chromatin, mất TAD boundary integrity, drift methylation) với epigenetic clock để hiểu và can thiệp vào lão hóa ở cấp độ mạng lưới. Liệu pháp partial reprogramming (kích hoạt ngắn hạn các TF Yamanaka để đảo chiều epigenetic aging) đang nhận được đầu tư lớn và đòi hỏi hiểu sâu về GRN trong lão hóa.
Clinical GRN models — tích hợp dữ liệu multi-omics single-cell từ bệnh nhân thực vào foundation models để cá nhân hóa GRN và từ đó dự đoán drug response. Mục tiêu dài hạn: với biopsy một bệnh nhân, chạy in silico perturbation trên GRN của bệnh nhân đó để chọn thuốc tối ưu — precision medicine thực sự ở cấp độ mạng lưới điều hòa.
Kết Luận
Gene regulatory network không phải là một đặc điểm thêm thắt của sinh học tế bào — nó là sinh học tế bào. Toàn bộ bản sắc tế bào, khả năng phản ứng với môi trường, và phương thức phát bệnh đều nằm trong kiến trúc của mạng lưới này. Từ phương trình Hill của toggle switch ODE, thuật toán KR normalization của Hi-C, đến DPI trong ARACNE hay rank-value tokenization của Geneformer — mỗi lớp kỹ thuật đều là một thấu kính nhìn vào GRN từ góc độ khác nhau.
Năm 2026, ba xu hướng hội tụ đang định hình lại lĩnh vực: (1) dữ liệu nhân quả quy mô lớn từ Perturb-seq atlas thay thế dần tương quan đơn thuần; (2) foundation models đa phương thức học đồng thời từ sequence, single-cell, và spatial omics; (3) ứng dụng lâm sàng trực tiếp đưa GRN vào pipeline dự đoán drug response cho từng bệnh nhân. Hiểu GRN không còn là mục tiêu cơ bản đơn thuần — nó đang trở thành hạ tầng của y học phân tử cá nhân hóa thế hệ tiếp theo.
Tài Liệu Tham Khảo
Lambert, S. A., Jolma, A., Campitelli, L. F., Das, P. K., Yin, Y., Albu, M., Chen, X., Taipale, J., Hughes, T. R., & Weirauch, M. T. (2018). The human transcription factors. Cell, 172(4), 650–665. https://doi.org/10.1016/j.cell.2018.01.029
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., & Greenleaf, W. J. (2013). Transposition of native chromatin for multimodal regulatory profiling and sequencing. Nature Methods, 10(12), 1213–1218. https://doi.org/10.1038/nmeth.2688
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., Amit, I., Lajoie, B. R., Sabo, P. J., Dorschner, M. O., Sandstrom, R., Bernstein, B., Bender, M. A., Groudine, M., Gnirke, A., Stamatoyannopoulos, J., Mirny, L. A., Lander, E. S., & Dekker, J. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326(5950), 289–293. https://doi.org/10.1126/science.1181369
Aibar, S., González-Blas, C. B., Moerman, T., Huynh-Thu, V. A., Imrichova, H., Hulselmans, G., Rambow, F., Marine, J. C., Geurts, P., Aerts, J., van den Oord, J., Atak, Z. K., Wouters, J., & Aerts, S. (2017). SCENIC: Single-cell regulatory network inference and clustering. Nature Methods, 14(11), 1083–1086. https://doi.org/10.1038/nmeth.4463
Kamimoto, K., Stringa, B., Hoffmann, C. M., Jindal, K., Solnica-Krezel, L., & Morris, S. A. (2023). Dissecting cell identity via network inference and in silico gene perturbation. Nature, 614(7949), 742–751. https://doi.org/10.1038/s41586-022-05688-9
Avsec, Ž., Agarwal, V., Visentin, D., Ledsam, J. R., Grabska-Barwinska, A., Taylor, K. R., Assael, Y., Jumper, J., Kohli, P., & Kelley, D. R. (2021). Effective gene expression prediction from sequence by integrating long-range interactions. Nature Methods, 18(10), 1196–1203. https://doi.org/10.1038/s41592-021-01252-x
Theodoris, C. V., Xiao, L., Chopra, A., Chaffin, M. D., Al Sayed, Z. R., Hill, M. C., Mantineo, H., Brydon, E. M., Zeng, Z., Liu, X. S., & Ellinor, P. T. (2023). Transfer learning enables predictions in network biology. Nature, 618(7965), 616–624. https://doi.org/10.1038/s41586-023-06139-9
La Manno, G., Soldatov, R., Zeisel, A., Braun, E., Hochgerner, H., Petukhov, V., Lidschreiber, K., Kastriti, M. E., Lönnerberg, P., Furlan, A., Fan, J., Borm, L. E., Liu, Z., van Bruggen, D., Guo, J., He, X., Barker, R., Sundström, E., Castelo-Branco, G., … Kharchenko, P. V. (2018). RNA velocity of single cells. Nature, 560(7719), 494–498. https://doi.org/10.1038/s41586-018-0414-6
Bergen, V., Lange, M., Peidli, S., Wolf, F. A., & Theis, F. J. (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nature Biotechnology, 38(12), 1408–1414. https://doi.org/10.1038/s41587-020-0591-3
Norman, T. M., Replogle, J. M., Broderick, K., Maier, M., Srinivasan, R., Bharat, S., & Weissman, J. S. (2019). Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science, 365(6455), 786–793. https://doi.org/10.1126/science.aax4438
Explore more like this
Từ Dữ Liệu Thô Đến Biến Thể Di Truyền: Genome Alignment, Variant Calling, QC và Annotation
Khi một mẫu DNA được đưa vào máy giải trình tự, kết quả trả về không phải là một câu trả lời hoàn chỉnh về bộ genome của cá thể...
Transcriptomics: Hành Trình Giải Mã Hệ Phiên Mã và Các Công Nghệ Chủ Chốt
Tế bào người chứa khoảng 3,2 tỷ cặp bazơ DNA — nhưng không phải tất cả đều được “đọc” cùng một lúc, hay ở mọi loại tế bào. Một tế...
Single-Cell Omics: Khi Khoa Học Nhìn Vào Từng Tế Bào
Hãy tưởng tượng bạn muốn hiểu một thành phố bằng cách hỏi một câu hỏi duy nhất với tất cả 10 triệu cư dân cùng lúc và lấy trung bình...
Comments