Transcriptomics: Hành Trình Giải Mã Hệ Phiên Mã và Các Công Nghệ Chủ Chốt

Tế bào người chứa khoảng 3,2 tỷ cặp bazơ DNA — nhưng không phải tất cả đều được “đọc” cùng một lúc, hay ở mọi loại tế bào. Một tế bào gan và một tế bào thần kinh mang cùng một genome, nhưng chúng trông và hành xử hoàn toàn khác nhau. Bí ẩn nằm ở lớp thông tin trung gian giữa DNA và protein: transcriptome, tập hợp tất cả các phân tử RNA được tổng hợp trong một tế bào tại một thời điểm nhất định. Transcriptomics là ngành khoa học nghiên cứu transcriptome này — ai được biểu hiện, bao nhiêu, ở đâu và khi nào. Trong bốn thập kỷ qua, từ những vệt băng điện di trên gel nitrocellulose đến các bản đồ gene expression ba chiều với độ phân giải đơn tế bào, transcriptomics đã trải qua một cuộc cách mạng kỹ thuật chưa từng có, mở ra những câu hỏi mà trước đây chúng ta thậm chí không biết cách đặt ra.
1. Transcriptome và Tầm Quan Trọng
1.1. Transcriptome Là Gì
Transcriptome được định nghĩa là toàn bộ tập hợp các phân tử RNA được phiên mã từ genome của một tế bào, mô hoặc sinh vật trong một điều kiện sinh lý cụ thể. Khác với genome vốn tương đối tĩnh và giống nhau giữa các tế bào trong cùng một sinh vật, transcriptome có tính động cao: nó thay đổi theo loại tế bào, giai đoạn phát triển, điều kiện môi trường, trạng thái bệnh lý và thậm chí theo từng giờ trong ngày.
Transcriptome không chỉ bao gồm mRNA (messenger RNA, RNA thông tin) mang mã cho protein. Nó còn bao trùm một bức tranh phong phú hơn nhiều: rRNA (ribosomal RNA), tRNA (transfer RNA), miRNA (microRNA), lncRNA (long non-coding RNA, RNA không mã hoá dài), snRNA (small nuclear RNA), snoRNA (small nucleolar RNA) và nhiều lớp RNA không mã hoá khác. Người ta ước tính rằng hơn 75% genome người được phiên mã thành RNA, nhưng chỉ khoảng 1,5–2% mã hoá cho protein. Phần lớn còn lại là RNA không mã hoá với vai trò điều hoà, cấu trúc và nhiều chức năng khác đang tiếp tục được khám phá.
Điều này dẫn đến một câu hỏi trung tâm của sinh học phân tử hiện đại: tại sao cùng một genome lại có thể cho ra các kiểu hình tế bào hoàn toàn khác nhau? Câu trả lời nằm ở điều hoà phiên mã (transcriptional regulation), cơ chế kiểm soát gene nào được bật, tắt hoặc điều chỉnh cường độ biểu hiện trong từng bối cảnh tế bào.
1.2. Tại Sao Nghiên Cứu Transcriptome
So với việc nghiên cứu genome, nghiên cứu transcriptome có những lợi thế đặc biệt. Genome cho biết một sinh vật có thể làm gì; transcriptome cho biết sinh vật đang thực sự đang làm gì tại thời điểm cụ thể đó. Về mặt ứng dụng lâm sàng, mức gene expression của một tập hợp gene thường phản ánh trạng thái bệnh tốt hơn so với trình tự DNA, vì nhiều bệnh lý không xuất phát từ đột biến gene mà từ sự rối loạn gene expression regulation.
Trong nghiên cứu ung thư, gene expression profile (gene expression profiling) đã biến đổi cách phân loại bệnh: một khối u vú với hình thái học giống nhau dưới kính hiển vi có thể chia thành bốn phân nhóm phân tử khác nhau, với tiên lượng và đáp ứng điều trị hoàn toàn khác biệt. Trong sinh học phát triển, transcriptome tiết lộ cách một tế bào trứng được thụ tinh đơn độc biệt hoá thành hàng trăm loại tế bào chuyên biệt. Trong sinh thái học, phân tích transcriptome môi trường (metatranscriptomics) cho phép nghiên cứu hoạt động chức năng của cộng đồng vi sinh vật mà không cần nuôi cấy.
1.3. Các Lớp RNA Chức Năng
Để hiểu transcriptomics, cần nắm rõ các lớp RNA chính:
| Loại RNA | Kích thước | Chức năng chính |
|---|---|---|
| mRNA | 200 bp – 10 kb | Mang thông tin mã hoá protein |
| rRNA | 120 nt – 5 kb | Thành phần cấu trúc và xúc tác của ribosome |
| tRNA | 70–90 nt | Vận chuyển amino acid trong dịch mã |
| miRNA | 18–23 nt | Điều hoà sau phiên mã, ức chế mRNA |
| lncRNA | > 200 nt | Điều hoà gene expression, cấu trúc nhiễm sắc thể |
| snRNA | 100–300 nt | Xử lý pre-mRNA (splicing) |
| circRNA | Vòng tròn | Bẫy miRNA, điều hoà biểu hiện |
| piRNA | 26–31 nt | Bảo vệ genome khỏi transposon ở tế bào mầm |
Bảng 1.1. Các lớp RNA chủ yếu trong tế bào và chức năng của chúng.
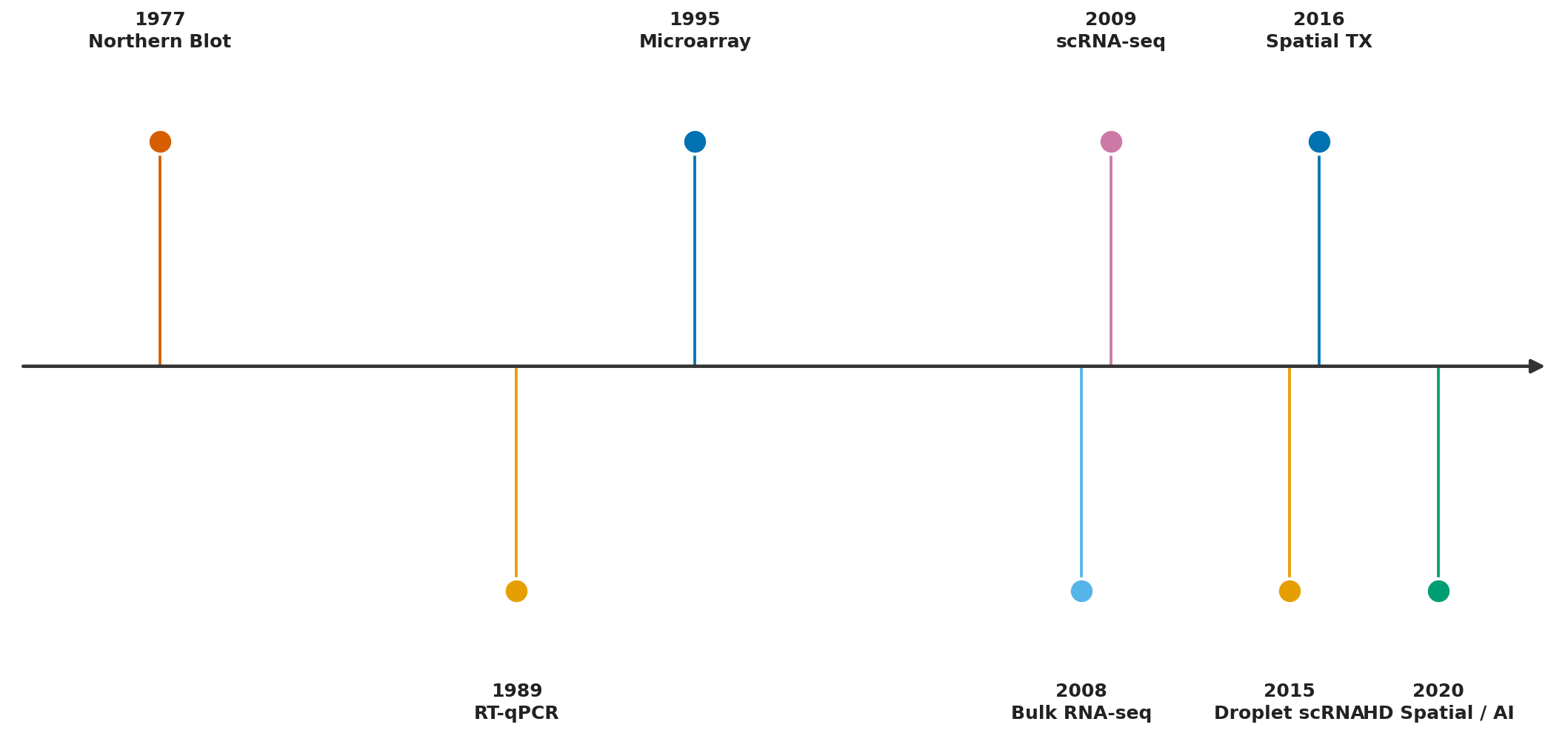
2. Dòng Thời Gian Công Nghệ
2.1. Thời Kỳ Tiền Kỹ Thuật Số (1977–1994)
Để đặt nền móng cho transcriptomics, các nhà khoa học cần một cách để phát hiện và đo lường RNA cụ thể trong hỗn hợp phức tạp của tế bào.
Northern Blot (lai phân tử RNA trên màng) là công nghệ đầu tiên cho phép phát hiện một RNA cụ thể trong mẫu sinh học. Được phát triển bởi Alwine, Kemp và Stark tại Stanford năm 1977, kỹ thuật này hoạt động theo nguyên tắc điện di RNA trên gel agarose, chuyển RNA lên màng nitrocellulose, rồi lai với một đầu dò nucleotide được đánh dấu phóng xạ bổ sung với trình tự mRNA đích. Kết quả là một vệt tối trên phim X-quang, với vị trí vệt xác định kích thước phân tử và độ đậm của vệt phản ánh mức độ biểu hiện. Northern Blot trả lời câu hỏi: “Gen X có được biểu hiện ở mô này không, và ở mức độ nào?” — nhưng chỉ cho một gene tại một thời điểm, đòi hỏi lượng mẫu lớn và mất nhiều ngày để thực hiện.
RT-PCR (Reverse Transcription Polymerase Chain Reaction, phản ứng chuỗi polymerase phiên mã ngược) được phát triển cuối thập niên 1980, kết hợp enzyme reverse transcriptase để chuyển mRNA thành cDNA, rồi khuếch đại bằng PCR. Phiên bản định lượng qRT-PCR (quantitative RT-PCR) cho phép đo mức gene expression với độ nhạy rất cao và khoảng tuyến tính rộng. Đây vẫn là “tiêu chuẩn vàng” để xác nhận kết quả gene expression cho đến ngày nay, dù bị giới hạn ở việc chỉ đo được một số ít gene mỗi thực nghiệm.
Trong giai đoạn này, nghiên cứu gene expression là một quá trình chậm chạp, giả thuyết-hướng dẫn: nhà khoa học phải biết trước muốn đo gene nào, rồi thiết kế thực nghiệm cho gene đó. Tư duy hệ thống, đo lường toàn bộ transcriptome cùng một lúc, vẫn còn là một giấc mơ xa vời.
2.2. Kỷ Nguyên Microarray (1995–2007)
Bước ngoặt đầu tiên đến năm 1995, khi Patrick Brown và Ron Davis tại Stanford công bố kỹ thuật DNA microarray (vi mảng DNA), còn gọi là gene chip (chip gene). Ý tưởng cốt lõi: thay vì dò tìm từng RNA một, tại sao không in hàng nghìn đầu dò DNA lên một tấm kính nhỏ và lai với toàn bộ mRNA từ mẫu thực nghiệm cùng một lúc?
Trong một thực nghiệm microarray điển hình, mRNA được chiết xuất từ tế bào, phiên mã ngược thành cDNA, rồi được đánh dấu bằng thuốc nhuộm huỳnh quang (thường dùng Cy3 màu xanh cho đối chứng và Cy5 màu đỏ cho mẫu thực nghiệm). Hai mẫu được lai đồng thời lên chip, và máy quét laser đọc cường độ huỳnh quang tại hàng chục nghìn điểm trên chip. Tỉ lệ cường độ đỏ/xanh tại mỗi điểm phản ánh mức biểu hiện tương đối của gene tương ứng (xem Hình 2.1).
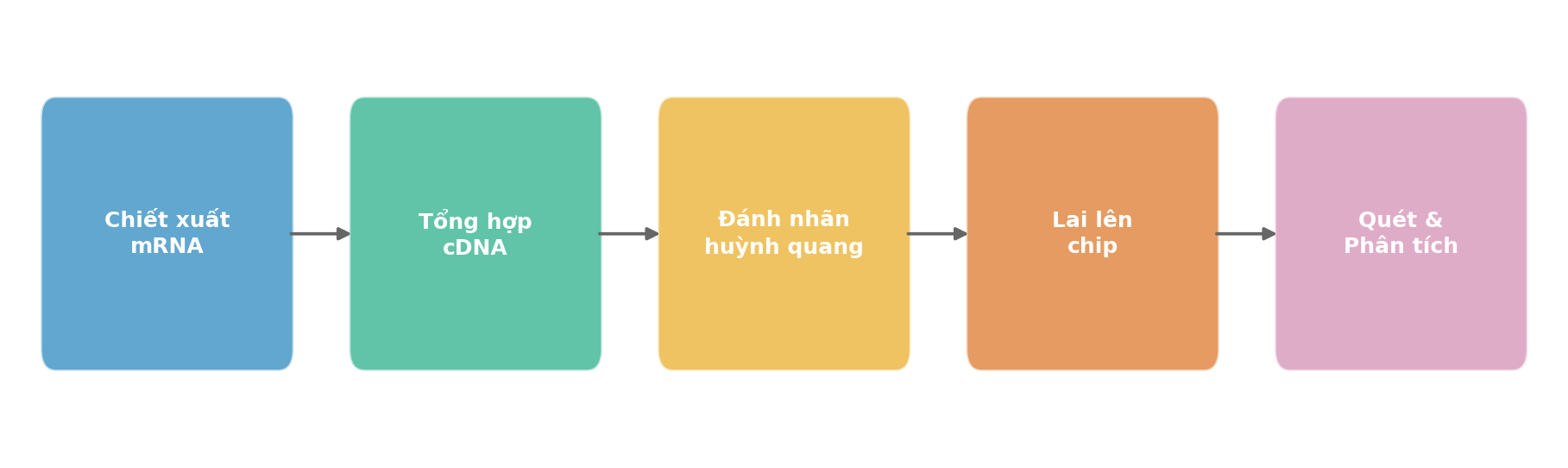 Hình 2.1. Quy trình tổng quát của một thực nghiệm DNA Microarray hai màu: từ chiết xuất mRNA, tổng hợp cDNA, đánh nhãn huỳnh quang (Cy3/Cy5), lai lên chip, đến quét laser và phân tích tỉ lệ biểu hiện.
Hình 2.1. Quy trình tổng quát của một thực nghiệm DNA Microarray hai màu: từ chiết xuất mRNA, tổng hợp cDNA, đánh nhãn huỳnh quang (Cy3/Cy5), lai lên chip, đến quét laser và phân tích tỉ lệ biểu hiện.
Nghiên cứu tiên phong của DeRisi et al. (1997) sử dụng microarray để phân tích gene expression của khoảng 6.000 gene nấm men, theo dõi diễn biến đáp ứng khi chuyển từ điều kiện sinh trưởng lên men sang hô hấp. Đây là lần đầu tiên toàn bộ transcriptome của một sinh vật được theo dõi trong một thực nghiệm duy nhất. Năm 2000, Perou et al. công bố phân loại phân tử ung thư vú dựa trên microarray, phát hiện ra ít nhất bốn phân nhóm phân tử khác biệt (Luminal A, Luminal B, HER2-enriched, Basal-like) với tiên lượng và đặc điểm lâm sàng riêng biệt. Công trình này đã thay đổi hoàn toàn cách chúng ta hiểu và điều trị ung thư vú.
Tuy nhiên, microarray có những hạn chế cơ bản không thể khắc phục. Nó chỉ đo được những RNA có trình tự đầu dò tương ứng thiết kế sẵn trên chip, nên không thể phát hiện transcript mới. Nó có tín hiệu nền cao do lai chéo phi đặc hiệu. Khoảng tuyến tính đo lường bị giới hạn. Và quan trọng hơn, nó không thể phân biệt các dạng ghép nối thay thế (alternative splicing) của cùng một gene.
2.3. Cách Mạng RNA Sequencing (2008–2015)
Năm 2008 đánh dấu một bước nhảy vọt khi hai nhóm nghiên cứu độc lập, Sultan et al. và Mortazavi et al., công bố phương pháp RNA-seq (RNA sequencing, giải trình tự RNA). Thay vì lai với đầu dò, RNA-seq chuyển đổi mRNA thành thư viện cDNA và giải trình tự trực tiếp bằng công nghệ thế hệ kế tiếp (NGS), thường là Illumina.
Quy trình RNA-seq cơ bản bao gồm: chiết xuất RNA, loại bỏ rRNA (vì rRNA chiếm 90–95% tổng RNA tế bào), phân mảnh RNA, phiên mã ngược tạo cDNA, thêm adapter, giải trình tự, và phân tích tin sinh học. Mỗi đoạn đọc (read) từ 50 đến 150 cặp bazơ được ánh xạ về genome tham chiếu, và số lượng read ánh xạ lên một gene phản ánh mức gene expression của gene đó.
RNA-seq có nhiều ưu điểm vượt trội so với microarray (xem Hình 2.2):
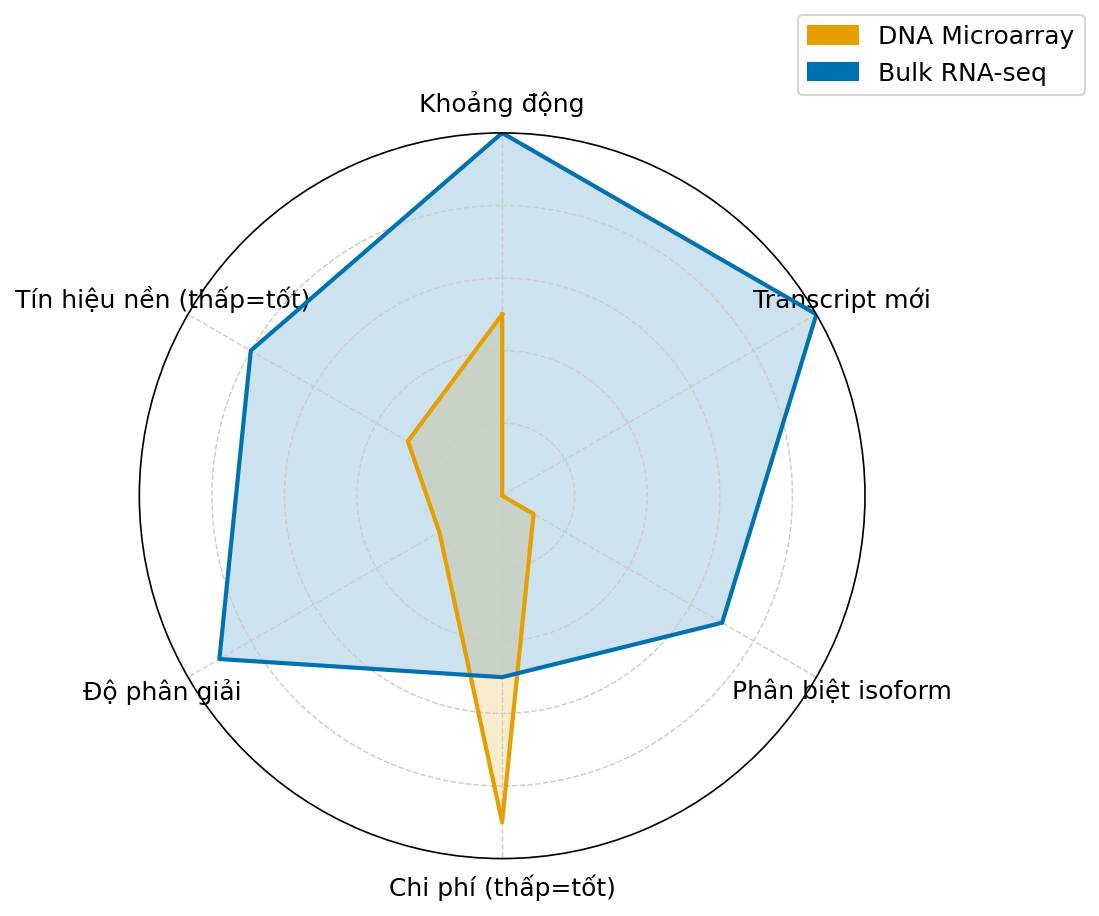 Hình 2.2. Biểu đồ radar so sánh khả năng kỹ thuật giữa DNA Microarray và Bulk RNA-seq trên sáu tiêu chí: khoảng động, phát hiện transcript mới, phân biệt isoform, chi phí, độ phân giải nucleotide và mức tín hiệu nền.
Hình 2.2. Biểu đồ radar so sánh khả năng kỹ thuật giữa DNA Microarray và Bulk RNA-seq trên sáu tiêu chí: khoảng động, phát hiện transcript mới, phân biệt isoform, chi phí, độ phân giải nucleotide và mức tín hiệu nền.
- Không giới hạn bởi thiết kế sẵn: phát hiện được transcript mới, đột biến, và gene fusion
- Khoảng động rộng: từ vài đến hàng triệu copy mRNA có thể được đo trong cùng một thực nghiệm
- Phân giải nucleotide: phân biệt được các dạng ghép nối thay thế, phát hiện SNP trong RNA
- Đo lường tuyệt đối: số lượng read ánh xạ có ý nghĩa tuyệt đối, không chỉ tương đối như trong microarray hybridisation
- Ít mẫu hơn: các giao thức RNA-seq hiện đại có thể hoạt động với dưới 10 ng RNA tổng số
Giai đoạn 2008–2015 chứng kiến RNA-seq thay thế microarray trong hầu hết các ứng dụng nghiên cứu, trở thành công cụ tiêu chuẩn cho phân tích gene expression. Đồng thời, tiến bộ trong tin sinh học đã tạo ra các công cụ phân tích như TopHat, HISAT2, STAR (alignment), Cufflinks, DESeq2, edgeR (phân tích differential expression) để xử lý dữ liệu quy mô lớn này.
2.4. Kỷ Nguyên Đơn Tế Bào (2009–Nay)
Dù RNA-seq đã cách mạng hoá nghiên cứu gene expression, nó vẫn có một hạn chế căn bản: nó đo lường transcriptome trung bình của hàng nghìn đến hàng triệu tế bào cùng một lúc. Trong một mô có nhiều loại tế bào, tín hiệu gene expression bị pha trộn và đồng nhất hoá. Tế bào ung thư bị che khuất bởi tế bào mô đệm xung quanh. Tế bào gốc hiếm gặp không thể phân biệt khỏi tế bào biệt hoá chiếm số đông.
Giải pháp đến năm 2009 khi Tang et al. công bố phương pháp single-cell RNA-seq (scRNA-seq, giải trình tự RNA đơn tế bào) đầu tiên, giải trình tự transcriptome của từng tế bào riêng lẻ từ phôi chuột. Về mặt nguyên tắc, scRNA-seq tương tự RNA-seq thông thường, nhưng thách thức kỹ thuật cực kỳ lớn: lượng RNA trong một tế bào đơn chỉ khoảng 10 picogram, đòi hỏi khuếch đại mạnh mẽ trước khi giải trình tự, và các artefact khuếch đại phải được kiểm soát cẩn thận.
Bước nhảy vọt lớn xảy ra năm 2015–2016 khi các công nghệ droplet-based (vi giọt) như 10x Genomics Chromium, inDrop và Drop-seq xuất hiện. Các công nghệ này đóng gói từng tế bào vào một giọt dầu cực nhỏ chứa hạt chuỗi với mã vạch phân tử (molecular barcode), cho phép giải trình tự hàng nghìn tế bào cùng một lúc trong một thực nghiệm duy nhất. Với 10x Genomics, chi phí giải trình tự mỗi tế bào giảm từ hàng trăm đô la xuống còn vài xu.
Các ứng dụng của scRNA-seq đã cách mạng hoá sinh học tế bào:
Bản đồ tế bào học (cell atlas): Dự án Human Cell Atlas, khởi động năm 2016, đặt mục tiêu lập bản đồ mọi loại tế bào trong cơ thể người bằng scRNA-seq. Đến nay, hàng chục triệu tế bào từ hầu hết các cơ quan đã được phân loại, phát hiện nhiều loại tế bào chưa từng được biết đến trước đây.
Phân tích quỹ đạo phát triển (trajectory analysis): Các công cụ như Monocle và Scanpy cho phép tái dựng quỹ đạo biệt hoá tế bào từ dữ liệu snapshot, tiết lộ con đường phát triển từ tế bào gốc đến tế bào trưởng thành.
Giải mã vi môi trường khối u (tumor microenvironment): scRNA-seq đã tiết lộ sự phức tạp đáng kinh ngạc của các loại tế bào miễn dịch và mô đệm trong khối u, chỉ ra những yếu tố dự đoán đáp ứng liệu pháp miễn dịch.
2.5. Chụp Cắt Lớp Không Gian (2016–Nay)
scRNA-seq giải quyết vấn đề phân giải tế bào, nhưng lại đánh đổi một thông tin quan trọng: vị trí không gian (spatial context). Khi tế bào được phân tán và giải trình tự riêng lẻ, chúng ta biết gene nào được biểu hiện trong tế bào đó, nhưng không biết tế bào đó đang ở đâu trong mô, cạnh tế bào nào, và thuộc vùng cấu trúc nào.
Spatial transcriptomics (phiên mã học không gian) ra đời để lấp đầy khoảng trống này. Các công nghệ này cho phép đo lường transcriptome trong khi vẫn giữ nguyên vị trí không gian của tế bào trong cấu trúc mô.
Năm 2016, Ståhl et al. công bố công nghệ Spatial Transcriptomics ban đầu (sau được mua lại và thương mại hoá bởi 10x Genomics thành Visium): các lát cắt mô được đặt lên mảng các điểm nắm bắt RNA (capture spots), mỗi điểm có đường kính 55 µm mang chuỗi mã vạch không gian duy nhất. mRNA khuếch tán xuống và gắn vào điểm nắm bắt tương ứng, rồi được giải trình tự. Kết quả là bản đồ gene expression gắn liền với toạ độ không gian trong mô.
Năm 2020–2022, nhiều công nghệ spatial với độ phân giải tế bào đơn xuất hiện: Stereo-seq (từ BGI), Slide-seq, MERFISH, seqFISH+ và Xenium (10x Genomics). MERFISH và seqFISH dùng kính hiển vi huỳnh quang đa vòng để phát hiện hàng nghìn gene trực tiếp trong tế bào mà không cần giải trình tự, đạt độ phân giải dưới tế bào (subcellular). Năm 2020, công nghệ spatial transcriptomics được tạp chí Nature Methods bình chọn là Phương pháp của Năm.
Những ứng dụng đầu tiên cho thấy sức mạnh của phương pháp này: mapping ranh giới vùng não chính xác đến từng lớp tế bào thần kinh, xác định domain vi môi trường khối u, và hiểu kiến trúc của trung tâm mầm trong hạch bạch huyết.
2.6. Giải Trình Tự Đọc Dài (2014–Nay)
Trong suốt giai đoạn RNA-seq và scRNA-seq, phần lớn dữ liệu đến từ công nghệ đọc ngắn của Illumina (50–150 bp). Đọc ngắn đặt ra thách thức cơ bản trong việc phân biệt các dạng ghép nối thay thế (alternative splicing): khi một pre-mRNA có thể được cắt ghép theo nhiều cách để tạo ra nhiều isoform protein khác nhau, việc tái tạo cấu trúc đầy đủ của từng isoform từ các mảnh ngắn giống như giải câu đố xếp hình với hàng nghìn mảnh trùng lặp.
Công nghệ đọc dài (long-read sequencing) từ PacBio (SMRT sequencing) và Oxford Nanopore Technologies (ONT) đã thay đổi điều này. PacBio có thể tạo ra các read 10–25 kb với độ chính xác đồng thuận (consensus accuracy) trên 99,9% khi dùng CCS (Circular Consensus Sequencing). Oxford Nanopore kết nối trực tiếp phân tử RNA mà không cần chuyển đổi thành DNA (direct RNA sequencing), loại bỏ artefact từ bước RT và khuếch đại, đồng thời phát hiện được các biến đổi hoá học của RNA (RNA modifications) như m6A methylation trực tiếp từ tín hiệu điện.
Trong transcriptomics, long-read sequencing cho phép tái tạo isoform transcriptome toàn phần, phát hiện các isoform mới và hiếm gặp, và nghiên cứu sự phối hợp ghép nối trên cùng một phân tử RNA duy nhất — điều không thể thực hiện với đọc ngắn.
3. Các Câu Hỏi Khoa Học Trung Tâm
3.1. Ai Biểu Hiện Gen Gì và Bao Nhiêu
Câu hỏi cơ bản nhất của transcriptomics là định lượng gene expression: trong điều kiện A và điều kiện B, gene nào biểu hiện cao hơn, thấp hơn, và mức độ chênh lệch là bao nhiêu? Khái niệm differential expression (differential expression) là nền tảng của hầu hết các phân tích transcriptomics.
Về mặt thống kê, phân tích differential expression đặt ra nhiều thách thức: dữ liệu đếm read tuân theo phân phối nhị thức âm (negative binomial), cần ước lượng phương sai từ số lượng mẫu lặp thường rất hạn chế (3–5 biological replicates), và cần điều chỉnh kiểm định bội (multiple testing correction) khi so sánh đồng thời hàng chục nghìn gene. Các gói R như DESeq2 và edgeR đã trở thành tiêu chuẩn vàng cho loại phân tích này, sử dụng mô hình thống kê tinh vi để giải quyết những thách thức trên.
3.2. Tế Bào Nào Đang Làm Gì
scRNA-seq đã chuyển đổi câu hỏi từ “mô này gene expression gì?” thành “loại tế bào nào trong mô này gene expression gì?”. Phân loại không giám sát (unsupervised clustering) dựa trên gene expression profile cho phép nhận dạng các loại tế bào và trạng thái tế bào mà không cần biết trước. Trong một mô não đơn giản, scRNA-seq có thể phân biệt hàng chục loại tế bào thần kinh với đặc điểm biểu hiện riêng biệt.
Bước tiếp theo là phân tích giao tiếp tế bào (cell-cell communication): các công cụ như CellChat và NicheNet suy luận từ dữ liệu scRNA-seq xem tế bào nào đang “nói chuyện” với tế bào nào thông qua tín hiệu ligand-receptor, mở ra cửa sổ quan sát vào mạng lưới tín hiệu phức tạp trong mô.
3.3. Transcript Nào Thực Sự Tạo Ra Protein
Mặc dù mRNA được phiên mã không đồng nghĩa với việc nó sẽ được dịch mã thành protein. Lớp thông tin tiếp theo — proteomics và translatome (hệ dịch mã) — đòi hỏi kỹ thuật riêng như Ribosome Profiling (Ribo-seq), giải trình tự các đoạn mRNA đang được ribosome dịch mã bảo vệ. Sự tích hợp giữa transcriptomics và proteomics là một lĩnh vực nghiên cứu đang phát triển nhanh chóng trong khuôn khổ multi-omics.
3.4. RNA Được Biến Đổi Hoá Học Như Thế Nào
Biến đổi hoá học của RNA (RNA modifications, epitranscriptomics) là lớp thông tin mới được khám phá. N6-methyladenosine (m6A), pseudouridine (Ψ), và hàng chục loại biến đổi khác ảnh hưởng đến sự ổn định, dịch mã và vị trí nội bào của mRNA. Các công nghệ như m6A-seq, Nanopore direct RNA sequencing và DART-seq cho phép đo lường biến đổi RNA ở quy mô transcriptome, mở ra lĩnh vực epitranscriptomics (thượng phiên mã học).
3.5. Hệ Phiên Mã Thay Đổi Theo Không Gian Như Thế Nào
Spatial transcriptomics đặt ra câu hỏi: gene expression thay đổi như thế nào dọc theo trục không gian trong mô? Kiến trúc vùng não, ranh giới khối u, gradient phát triển phôi — tất cả đều phản ánh sự thay đổi có quy luật của transcriptome trong không gian ba chiều. Hiểu được những gradient này là chìa khoá để tái dựng cách các tín hiệu phát triển định hướng sự biệt hoá tế bào theo vị trí trong cơ thể.
4. Sự Tiến Hoá Công Nghệ và Các Xu Hướng Hiện Đại
4.1. Từ Quần Thể Đến Đơn Tế Bào Đến Đơn Phân Tử
Sự tiến hoá của transcriptomics có thể được hiểu qua ba trục nâng cao độ phân giải (xem Hình 4.1):
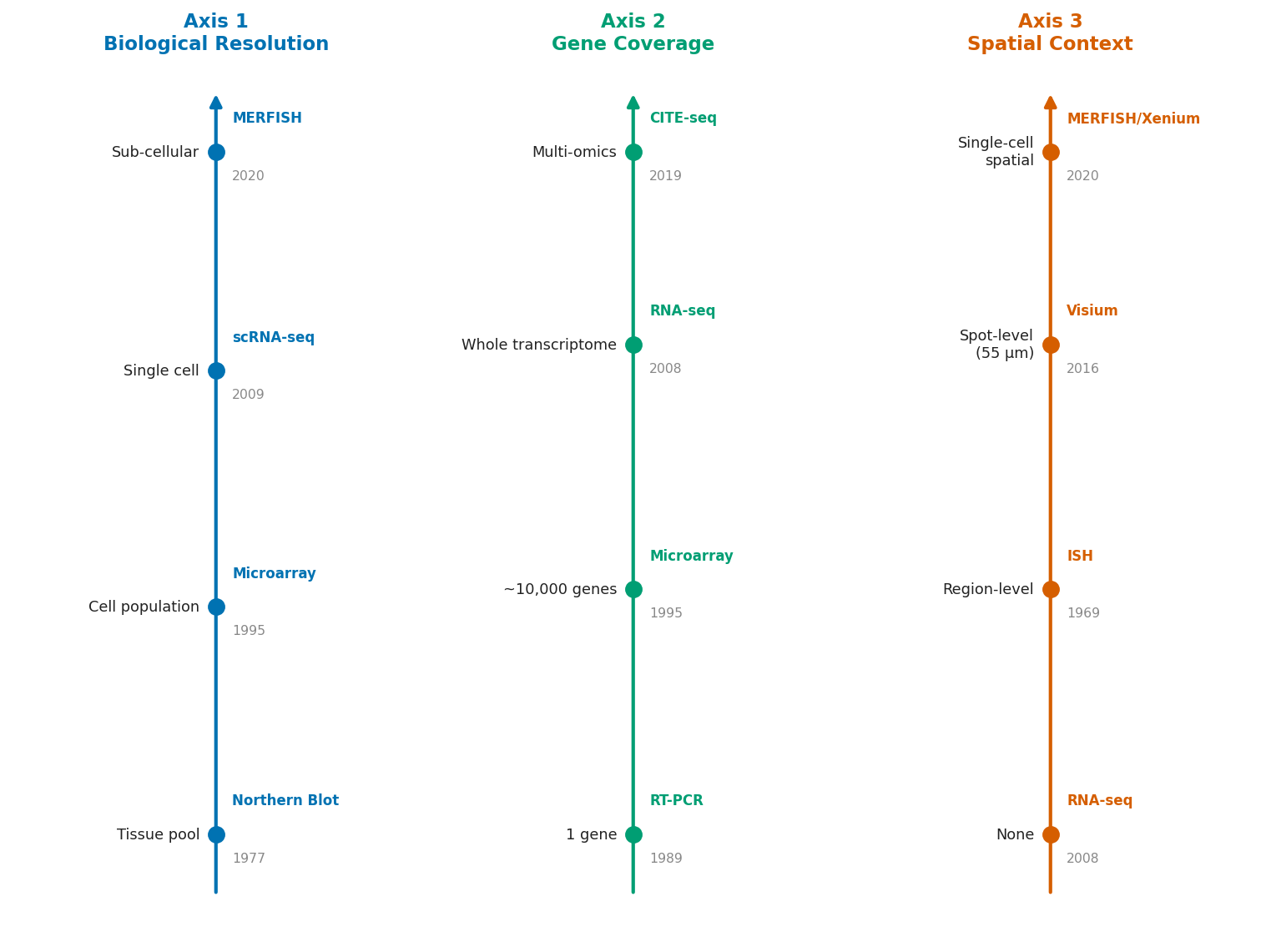 Hình 4.1. Ba trục tiến hoá công nghệ transcriptomics: độ phân giải sinh học (từ mô đến tế bào đơn), số gene đo đồng thời (từ 1 gene đến toàn transcriptome), và thông tin không gian (từ không có đến ba chiều).
Hình 4.1. Ba trục tiến hoá công nghệ transcriptomics: độ phân giải sinh học (từ mô đến tế bào đơn), số gene đo đồng thời (từ 1 gene đến toàn transcriptome), và thông tin không gian (từ không có đến ba chiều).
Trục #1: Độ phân giải sinh học. Đo lường di chuyển từ mức mô (bulk), xuống mức tế bào đơn (single-cell), và đang hướng tới mức phân tử đơn (single-molecule) hay thậm chí dưới tế bào (subcellular).
Trục #2: Số lượng gene đo đồng thời. Từ 1 gene (Northern Blot, RT-PCR), đến hàng nghìn gene (microarray), đến toàn bộ transcriptome (RNA-seq), đến đa omics đồng thời (multi-omics single-cell).
Trục #3: Thông tin ngữ cảnh. Từ không có ngữ cảnh không gian (RNA-seq, scRNA-seq), đến thông tin không gian 2D (spatial transcriptomics vi điểm), đến không gian 3D toàn phần (volumetric spatial transcriptomics).
4.2. Tích Hợp Đa Omics
Không một lớp omics nào có thể cung cấp bức tranh đầy đủ về tế bào. Xu hướng hiện đại là tích hợp đa omics (multi-omics integration), đo lường nhiều lớp thông tin từ cùng một tế bào hoặc cùng một mẫu sinh học.
Các phương pháp đồng thời đo scRNA-seq và ATAC-seq (chromatin accessibility, khả năng tiếp cận nhiễm sắc thể) như 10x Multiome, hay đo RNA và protein bề mặt như CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing), cho phép liên kết trực tiếp gene expression với trạng thái chromatin và kiểu hình protein trong cùng một tế bào. Điều này mở ra khả năng hiểu cơ chế gene expression regulation ở cấp độ tế bào đơn.
4.3. Giải Trình Tự Không Khuếch Đại
Một hạn chế của tất cả các phương pháp RNA-seq hiện tại là bước khuếch đại PCR, vốn gây ra thiên lệch (amplification bias) và làm mất thông tin về số lượng phân tử RNA ban đầu (copy number). Giải pháp là phân tử đồng nhất duy nhất (Unique Molecular Identifiers, UMIs): gắn mã vạch phân tử ngẫu nhiên vào mỗi phân tử mRNA trước khi khuếch đại, cho phép đếm số phân tử ban đầu thay vì số read sau khuếch đại. UMI hiện là tiêu chuẩn trong hầu hết các giao thức scRNA-seq.
Tương lai xa hơn là giải trình tự không khuếch đại (amplification-free sequencing): Nanopore direct RNA sequencing (dRNA-seq) đọc trực tiếp phân tử RNA mà không cần RT hay khuếch đại, với thông tin về biến đổi nucleotide và methylation đi kèm, nhưng vẫn đang trong giai đoạn phát triển để đạt độ chính xác cần thiết cho phân tích differential expression.
4.4. Trí Tuệ Nhân Tạo trong Transcriptomics
Sự bùng nổ của dữ liệu transcriptomics đã tạo ra nhu cầu cấp thiết về các phương pháp phân tích mới. Học sâu (deep learning) và mô hình nền tảng ngôn ngữ lớn cho sinh học (foundation models) đang được áp dụng vào transcriptomics theo nhiều hướng.
scGPT và Geneformer là các mô hình transformer được huấn luyện trước trên hàng chục đến hàng trăm triệu tế bào từ các tập dữ liệu scRNA-seq lớn. Tương tự như GPT trong xử lý ngôn ngữ tự nhiên, các mô hình này học được “ngôn ngữ” của gene expression và có thể được tinh chỉnh (fine-tuned) cho nhiều tác vụ hạ nguồn: phân loại tế bào, dự đoán đáp ứng thuốc, suy luận mạng điều hoà gene. Đây là một trong những hướng nghiên cứu sôi động nhất trong tin sinh học hiện đại.
5. Thách Thức và Hướng Phát Triển
5.1. Thách Thức Kỹ Thuật
Dropout trong scRNA-seq: Do lượng RNA trong đơn tế bào rất thấp, nhiều gene biểu hiện thấp không được phát hiện trong một số tế bào nhất định, tạo ra các giá trị không về kỹ thuật (technical zeros). Phân biệt dropout kỹ thuật với sự vắng mặt sinh học thực sự là một bài toán giải thống kê chưa hoàn toàn được giải quyết.
Batch effects: Khi dữ liệu từ nhiều phòng thí nghiệm, thời điểm thực nghiệm hay nền tảng công nghệ khác nhau được tích hợp, hiệu ứng lô (batch effects) có thể che khuất tín hiệu sinh học thực. Các phương pháp hiệu chỉnh batch như Harmony, BBKNN và scVI dùng mô hình xác suất để loại bỏ biến thiên kỹ thuật trong khi bảo tồn biến thiên sinh học.
Độ phân giải không gian: Công nghệ spatial transcriptomics thế hệ đầu như Visium có độ phân giải mỗi capture spot 55 µm, chứa nhiều tế bào. Các công nghệ phân giải đơn tế bào như MERFISH và Xenium hạn chế ở số lượng gene đo được (vài nghìn so với toàn bộ transcriptome của RNA-seq). Đây là đánh đổi giữa độ phủ transcriptome và độ phân giải không gian vẫn đang được giải quyết.
5.2. Thách Thức Của Dữ Liệu Lớn
Một thực nghiệm scRNA-seq thông thường tạo ra dữ liệu cho 10.000 đến 50.000 tế bào, mỗi tế bào có vector biểu hiện 30.000 gene. Đây là ma trận khổng lồ, thưa thớt và nhiễu. Lưu trữ, xử lý và phân tích những tập dữ liệu này đòi hỏi cơ sở hạ tầng tính toán đáng kể và các thuật toán giảm chiều và phân cụm hiệu quả như PCA, UMAP (Uniform Manifold Approximation and Projection) và t-SNE.
Thách thức về khả năng tái tạo (reproducibility) cũng rất nghiêm trọng trong lĩnh vực này: các kết quả phân tích scRNA-seq có thể thay đổi đáng kể tùy thuộc vào lựa chọn tham số trong nhiều bước của quy trình phân tích, từ ngưỡng lọc tế bào đến số lượng chiều chính thành phần đến thuật toán phân cụm. Chuẩn hoá và đánh giá quy trình phân tích là một lĩnh vực nghiên cứu phương pháp học đang phát triển.
5.3. Hướng Tương Lai
Transcriptomics thời gian thực: Kỹ thuật live-cell transcriptomics đo gene expression trong tế bào sống theo thời gian thực, kết hợp kính hiển vi huỳnh quang với các cảm biến RNA (MS2 system, CRISPRa-based reporters) để theo dõi động học gene expression mà không cần giết chết tế bào.
Tích hợp dữ liệu đa chiều: Tích hợp scRNA-seq, spatial transcriptomics, ATAC-seq, proteomics và dữ liệu hình ảnh trong một mô hình thống nhất sẽ cho phép hiểu đầy đủ về trạng thái tế bào ở tất cả các lớp thông tin sinh học.
Y học chính xác dựa trên transcriptomics: Gene expression profile từ tumor biopsy sample và tế bào tuần hoàn ngày càng được dùng để hướng dẫn điều trị cá nhân hoá, dự đoán đáp ứng liệu pháp miễn dịch và phát hiện kháng thuốc sớm.
6. So Sánh Tổng Hợp Các Công Nghệ
| Công nghệ | Năm | Độ phủ gene | Độ phân giải | Thông tin không gian | Ưu điểm nổi bật |
|---|---|---|---|---|---|
| Northern Blot | 1977 | Một gene | Mô/Tế bào | Không | Xác nhận kích thước RNA |
| RT-qPCR | 1989+ | Vài gene | Mô/Tế bào | Không | Độ nhạy cao, tiêu chuẩn vàng |
| Microarray | 1995 | Hàng nghìn gene | Mô | Không | Throughput cao lần đầu |
| EST sequencing | 1991–2005 | Một phần transcriptome | Mô | Không | Phát hiện transcript mới |
| Bulk RNA-seq | 2008 | Toàn transcriptome | Mô | Không | Khoảng động rộng, phát hiện isoform |
| scRNA-seq | 2009+ | Toàn transcriptome | Tế bào đơn | Không | Phân loại tế bào, trajectories |
| Spatial TX | 2016+ | Toàn TXome/Panel | Vùng mô/Tế bào | Có (2D/3D) | Ngữ cảnh không gian |
| Long-read RNA | 2014+ | Toàn transcriptome | Mô/Tế bào | Không | Phân giải isoform, RNA mods |
| Direct RNA (ONT) | 2017+ | Toàn transcriptome | Mô/Tế bào | Không | Biến đổi RNA, không RT |
Bảng 6.1. So sánh tổng hợp các công nghệ transcriptomics theo chiều lịch sử và kỹ thuật.
Kết Luận
Transcriptomics đã đi một hành trình phi thường trong gần năm thập kỷ: từ những vệt băng đơn độc trên tấm màng nitrocellulose đến bản đồ gene expression không gian ba chiều với độ phân giải đơn tế bào. Mỗi bước nhảy công nghệ không chỉ nâng cao độ nhạy và throughput, mà còn mở ra những câu hỏi sinh học hoàn toàn mới mà thế hệ công nghệ trước không thể đặt ra.
Những gì còn lại là sự tổng hợp: không có công nghệ nào đứng một mình. Northern Blot vẫn được dùng để xác nhận. qRT-PCR vẫn là tiêu chuẩn vàng. Bulk RNA-seq cho phân tích quần thể lớn. scRNA-seq giải mã sự đa dạng tế bào. Spatial transcriptomics đặt thông tin vào ngữ cảnh không gian. Long-read RNA giải quyết isoform. Và tất cả đang hội tụ về hướng tích hợp đa omics trong tế bào đơn với thông tin không gian.
Transcriptomics ngày nay không còn là ngành nghiên cứu thuần tuý của sinh học phân tử: nó là giao thoa của sinh học, vật lý, khoa học máy tính và thống kê, tạo ra một lĩnh vực khoa học dữ liệu sinh học của thế kỷ 21. Trong bài tutorial riêng, chúng tôi sẽ hướng dẫn quy trình phân tích thực hành từ dữ liệu bulk RNA-seq và scRNA-seq, từ bước xử lý dữ liệu thô đến phân tích differential expression và trực quan hoá kết quả.
Tài Liệu Tham Khảo
Alwine, J. C., Kemp, D. J., & Stark, G. R. (1977). Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proceedings of the National Academy of Sciences, 74(12), 5350–5354. https://doi.org/10.1073/pnas.74.12.5350
Schena, M., Shalon, D., Davis, R. W., & Brown, P. O. (1995). Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science, 270(5235), 467–470. https://doi.org/10.1126/science.270.5235.467
Perou, C. M., Sørlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., Pollack, J. R., Ross, D. T., Johnsen, H., Akslen, L. A., Fluge, Ø., Pergamenschikov, A., Williams, C., Zhu, S. X., Lønning, P. E., Børresen-Dale, A. L., Brown, P. O., & Botstein, D. (2000). Molecular portraits of human breast tumours. Nature, 406(6797), 747–752. https://doi.org/10.1038/35021093
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., & Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, 5(7), 621–628. https://doi.org/10.1038/nmeth.1226
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B., Siddiqui, A., Luo, K., & Surani, M. A. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods, 6(5), 377–382. https://doi.org/10.1038/nmeth.1315
Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., Trombetta, J. J., Weitz, D. A., Sanes, J. R., Shalek, A. K., Regev, A., & McCarroll, S. A. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Ståhl, P. L., Salmén, F., Vickovic, S., Lundmark, A., Navarro, J. F., Magnusson, J., Giacomello, S., Asp, M., Westholm, J. O., Huss, M., Mollbrink, A., Linnarsson, S., Codeluppi, S., Borg, Å., Pontén, F., Costea, P. I., Sahlén, P., Mulder, J., Bergmann, O., Bhatt, D. L., … Frisén, J. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 353(6294), 78–82. https://doi.org/10.1126/science.aaf2403
Stark, R., Grzelak, M., & Hadfield, J. (2019). RNA sequencing: The teenage years. Nature Reviews Genetics, 20(11), 631–656. https://doi.org/10.1038/s41576-019-0150-2
Haque, A., Engel, J., Teichmann, S. A., & Lönnberg, T. (2017). A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Medicine, 9(1), 75. https://doi.org/10.1186/s13073-017-0467-4
Moses, L., & Pachter, L. (2022). Museum of spatial transcriptomics. Nature Methods, 19(5), 534–546. https://doi.org/10.1038/s41592-022-01409-2
Garalde, D. R., Snell, E. A., Jachimowicz, D., Sipos, B., Lloyd, J. H., Bruce, M., Pantic, N., Admassu, T., James, P., Warland, A., Jordan, M., Ciccone, J., Serra, S., Keenan, J., Martin, S., McNeill, L., Wallace, E. J., Jayasinghe, L., Wright, C., … Turner, D. J. (2018). Highly parallel direct RNA sequencing on an array of nanopores. Nature Methods, 15(3), 201–206. https://doi.org/10.1038/nmeth.4577
Explore more like this
Gene Regulatory Network: Từ Phân Tử Đến Multi-Omics Và Ứng Dụng Lâm Sàng
Genome của người chứa khoảng 20.000 gene mã hóa protein, nhưng không phải gene nào cũng hoạt động ở mọi tế bào trong mọi thời điểm. Một tế bào gan,...
Từ Dữ Liệu Thô Đến Biến Thể Di Truyền: Genome Alignment, Variant Calling, QC và Annotation
Khi một mẫu DNA được đưa vào máy giải trình tự, kết quả trả về không phải là một câu trả lời hoàn chỉnh về bộ genome của cá thể...
Single-Cell Omics: Khi Khoa Học Nhìn Vào Từng Tế Bào
Hãy tưởng tượng bạn muốn hiểu một thành phố bằng cách hỏi một câu hỏi duy nhất với tất cả 10 triệu cư dân cùng lúc và lấy trung bình...
Comments