Single-Cell Omics: Khi Khoa Học Nhìn Vào Từng Tế Bào

Hãy tưởng tượng bạn muốn hiểu một thành phố bằng cách hỏi một câu hỏi duy nhất với tất cả 10 triệu cư dân cùng lúc và lấy trung bình câu trả lời của họ. Bạn sẽ nhận được một con số mô tả “người dân trung bình” — không giàu không nghèo, không già không trẻ, không vui không buồn — một thực thể thống kê không tồn tại trong thực tế. Nhưng đó chính xác là những gì omics truyền thống đã làm với tế bào trong hơn hai thập kỷ: một mẫu RNA-seq bulk phân tích hàng triệu tế bào cùng lúc và trả về mức biểu hiện gen “trung bình” của cả quần thể, che khuất hoàn toàn sự đa dạng sinh học bên trong.
Single-cell omics (omics đơn tế bào: nhóm công nghệ đo lường các lớp phân tử sinh học — transcriptome, epigenome, proteome, genome — ở độ phân giải từng tế bào riêng lẻ) đã thay đổi quy tắc này từ gốc rễ. Kể từ khi các nền tảng droplet-based đầu tiên xuất hiện vào năm 2015, single-cell omics đã cho phép chúng ta vẽ bản đồ kiểu tế bào của mọi cơ quan trong cơ thể người, giải phẫu hệ sinh thái phức tạp của khối u ở độ phân giải tế bào đơn, và khám phá những trạng thái tế bào trung gian mà không kỹ thuật nào trước đó có thể nhìn thấy. Bài viết này trình bày toàn cảnh của lĩnh vực — từ nguyên lý kỹ thuật đến các nghiên cứu nền tảng đã định hình lại sinh học hiện đại, để bạn đọc có thể xây dựng trực giác vững chắc về một trong những lĩnh vực phát triển nhanh nhất của khoa học sự sống.
1. Vấn Đề Nền Tảng
1.1. Nghịch Lý Của Phép Đo Quần Thể
Khi các nhà khoa học phân tích mô não bằng RNA-seq bulk, họ nhận được một danh sách gene với mức biểu hiện trung bình. Nhưng não chứa hàng trăm loại tế bào khác nhau — tế bào thần kinh (neurons), tế bào đệm (astrocytes), tế bào vi thần kinh đệm (microglia), tế bào nội mô mạch máu — mỗi loại biểu hiện một chương trình gen hoàn toàn khác nhau. Mức “trung bình” của tất cả các loại tế bào này không mô tả bất kỳ tế bào thực sự nào trong não.
Vấn đề trở nên đặc biệt nghiêm trọng trong nghiên cứu ung thư. Một khối u không chỉ gồm tế bào ung thư — nó là một hệ sinh thái phức tạp (tumor microenvironment, TME: vi môi trường khối u bao gồm tế bào ung thư, tế bào miễn dịch xâm nhập, tế bào sợi hoạt hóa, và tế bào nội mô mạch máu, tương tác với nhau để tạo ra đặc tính kháng thuốc và tiến triển ung thư). Khi RNA-seq bulk đo toàn bộ khối u, tín hiệu của mỗi thành phần bị hòa tan trong tín hiệu hỗn hợp. Một gene biểu hiện cao — liệu điều đó có nghĩa là gene đó tăng biểu hiện trong tế bào ung thư, hay chỉ vì tỷ lệ tế bào miễn dịch trong mẫu cao hơn? Câu hỏi này không thể trả lời từ dữ liệu bulk.
Tính dị chất tế bào (cellular heterogeneity: sự khác biệt về gene expression, trạng thái chức năng, và đặc điểm phân tử giữa các tế bào trong cùng một mô hay quần thể — kể cả trong cùng một “loại tế bào” theo định nghĩa truyền thống) chính là thực tế sinh học căn bản mà đo lường quần thể không thể tiếp cận được.
1.2. Giải Pháp Đơn Tế Bào
Câu trả lời logic là: đo từng tế bào riêng lẻ. Nhưng tế bào người rất nhỏ (10–20 micromet), chứa lượng RNA rất ít (khoảng 10–30 picogram), và một mô thực sự chứa đến hàng triệu tế bào. Thách thức kỹ thuật là phải tách, gán nhãn, và đo lường hàng nghìn đến hàng chục nghìn tế bào riêng lẻ trong cùng một thực nghiệm với độ nhạy đủ cao để phát hiện biểu hiện của hàng nghìn gene trong lượng RNA cực nhỏ từ một tế bào.
Bước đột phá cốt lõi đến từ hai hướng kỹ thuật hội tụ vào khoảng năm 2015: vi lưu chất (microfluidics: kỹ thuật thao tác chất lỏng ở quy mô cực nhỏ, từ nanoliter đến picoliter, cho phép xử lý hàng nghìn tế bào đơn trong một thiết bị nhỏ gọn) và barcode phân tử (molecular barcoding: gán một chuỗi DNA ngẫu nhiên duy nhất — cell barcode — cho từng tế bào và một chuỗi UMI — Unique Molecular Identifier — cho từng phân tử RNA, cho phép truy ngược về nguồn gốc tế bào sau khi giải trình tự và khử sai số khuếch đại PCR). Hai công nghệ này kết hợp tạo ra nền tảng của cuộc cách mạng đơn tế bào.
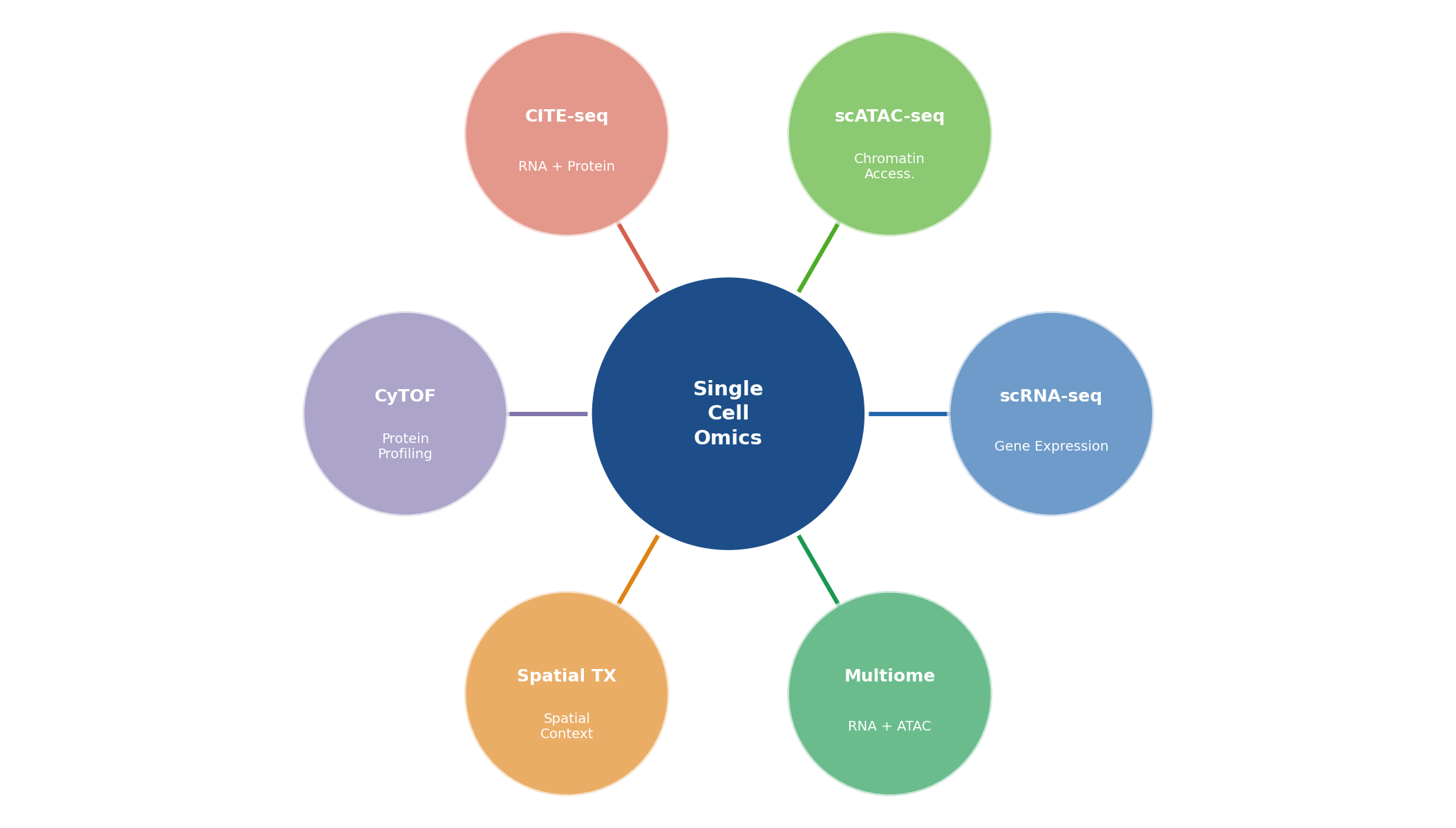
Hình 1.1. Bản đồ các công nghệ single-cell omics chính: scRNA-seq, scATAC-seq, CITE-seq, CyTOF, Spatial Transcriptomics, và Multiome — mỗi công nghệ đo một lớp thông tin phân tử khác nhau từ cùng một tế bào đơn.
2. scRNA-seq
2.1. Nguyên Lý Kỹ Thuật
scRNA-seq (single-cell RNA sequencing: giải trình tự RNA đơn tế bào — đo gene expression của từng tế bào riêng lẻ thay vì lấy trung bình của cả quần thể) là công nghệ nền tảng và phổ biến nhất trong single-cell omics. Trong thiết kế droplet-based, dung dịch chứa tế bào và dung dịch chứa hạt bead (hạt polymer nhỏ mang hàng nghìn trình tự oligo-dT poly-A với cell barcode và UMI gắn sẵn) được bơm qua hai kênh vi lưu chất hội tụ với dầu không trộn lẫn. Tại điểm hội tụ, mỗi giọt dầu siêu nhỏ bẫy — theo xác suất Poisson — lý tưởng một tế bào và một bead. Tế bào vỡ ra trong giọt, giải phóng mRNA, và enzyme reverse transcriptase (phiên mã ngược: enzyme tổng hợp cDNA từ khuôn mRNA) trên bead chuyển đổi mRNA thành cDNA gắn với barcode và UMI của bead đó. Từ đây, thư viện cDNA từ hàng nghìn giọt được giải trình tự cùng nhau và barcode là cơ chế để tách từng tế bào sau giải trình tự.
(xem Hình 2.1 cho quy trình tổng quát)
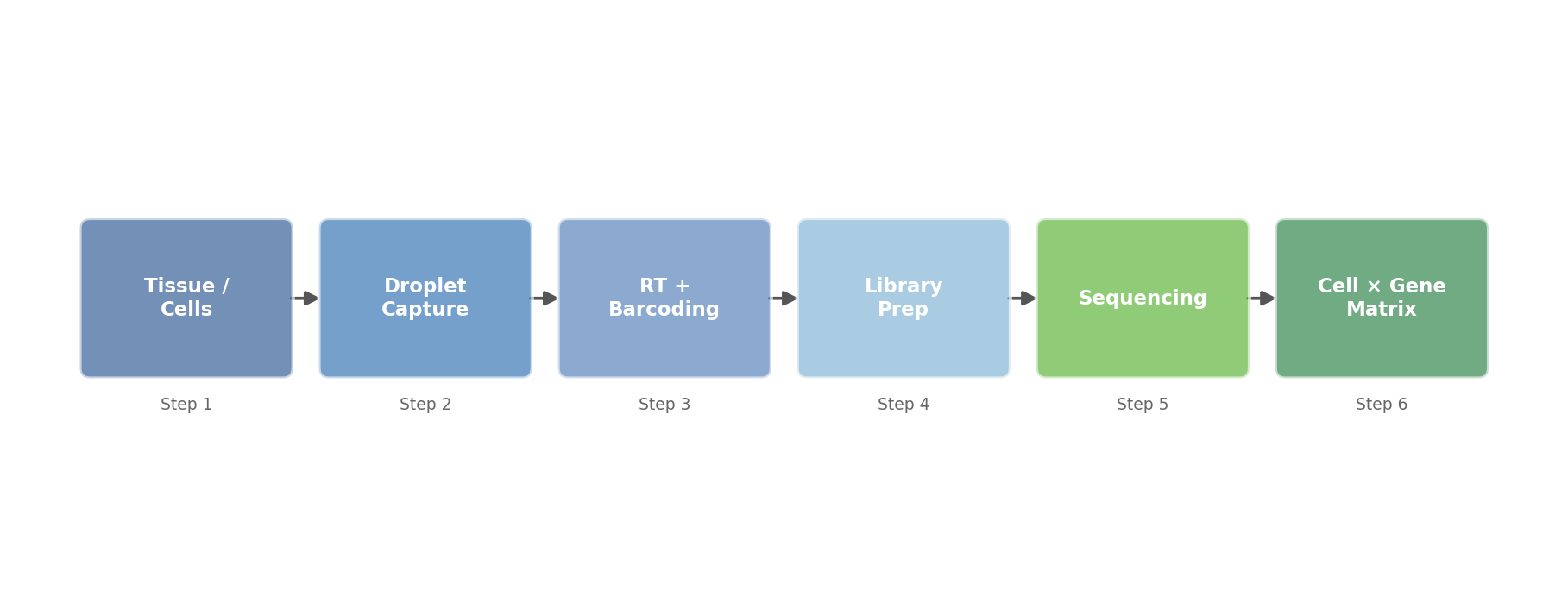 Hình 2.1. Quy trình scRNA-seq droplet-based: từ hỗn hợp tế bào qua bẫy droplet microfluidics và barcoding, đến giải trình tự và phân tích UMAP.
Hình 2.1. Quy trình scRNA-seq droplet-based: từ hỗn hợp tế bào qua bẫy droplet microfluidics và barcoding, đến giải trình tự và phân tích UMAP.
2.2. Các Nền Tảng Chính
Drop-seq (Macosko et al., 2015) và inDrop (Klein et al., 2015) là hai hệ thống droplet-based đầu tiên, công bố gần như đồng thời và nhanh chóng chứng minh rằng hàng nghìn tế bào retina chuột có thể phân tích cùng một lúc, tiết lộ cả các loại tế bào đã biết và kiểu tế bào mới chưa từng được mô tả trước đó. Năm 2017, 10x Chromium (Zheng et al., 2017) thương mại hóa công nghệ với quy trình chuẩn hóa, cho phép profiling 68.000 tế bào máu ngoại vi trong một thực nghiệm — minh chứng đầu tiên về khả năng áp dụng ở quy mô quần thể lớn.
Song song, Smart-seq2 (Picelli et al., 2013) theo hướng khác: mỗi tế bào được phân lập riêng lẻ bằng FACS vào từng giếng của plate 96 hoặc 384. Phương pháp này tốn kém hơn và xử lý ít tế bào hơn, nhưng cho độ bao phủ transcriptome cao hơn và khả năng phân tích splice variant (dạng ghép nối thau thế: các isoform RNA khác nhau của cùng một gene được tạo ra qua alternative splicing) — rất hữu ích khi cần phân tích sâu một số ít tế bào quý hiếm.
| Nền tảng | Throughput | Độ nhạy/tế bào | Chi phí/tế bào | Đặc điểm riêng |
|---|---|---|---|---|
| 10x Chromium | Rất cao (10k–50k) | Trung bình (~3k gene/tế bào) | Thấp | Chuẩn công nghiệp |
| Drop-seq | Cao (5k–30k) | Trung bình | Thấp | Open-source |
| Smart-seq2 | Thấp (96–384) | Cao (~8k gene/tế bào) | Cao | Full-length, isoform-aware |
| Parse Biosciences | Rất cao (>100k) | Trung bình | Rất thấp | Không cần thiết bị đặc biệt |
Bảng 2.1. So sánh các nền tảng scRNA-seq chính về throughput, độ nhạy, chi phí và đặc điểm kỹ thuật.
2.3. Phân Tích Dữ Liệu và UMAP
Kết quả của một thực nghiệm scRNA-seq là một ma trận biểu hiện đơn tế bào (single-cell expression matrix: ma trận số với hàng là tế bào và cột là gene, giá trị tại mỗi ô là UMI count của gene đó trong tế bào đó — rất thưa thớt vì mỗi tế bào chỉ biểu hiện một phần genome). Ma trận này thường có kích thước hàng chục nghìn tế bào × hai mươi nghìn gene.
Để tìm cấu trúc từ ma trận khổng lồ này, phân tích đi qua một chuỗi bước chuẩn: lọc tế bào chất lượng thấp → chuẩn hóa → giảm chiều bằng PCA (Principal Component Analysis: phân tích thành phần chính, giảm từ 20.000 chiều gene xuống còn 30–50 chiều chính) → phân cụm bằng thuật toán graph-based (Louvain hoặc Leiden) → trực quan hóa bằng UMAP (Uniform Manifold Approximation and Projection: phương pháp giảm chiều không tuyến tính tạo ra bản đồ 2D trong đó các tế bào tương tự nhau về gene expression xuất hiện gần nhau, phản ánh cấu trúc tuyến hệ phát triển và ranh giới giữa các loại tế bào). Trên bản đồ UMAP, mỗi điểm là một tế bào, màu sắc biểu thị loại tế bào đã được chú giải, và khoảng cách phản ánh sự tương đồng phân tử.
Điều quan trọng cần hiểu về UMAP: không gian 2D là một projection (hình chiếu) của không gian nhiều chiều thực sự. Khoảng cách tuyệt đối giữa các cụm trên UMAP không có ý nghĩa vật lý, nhưng cấu trúc cục bộ — hai tế bào ở gần nhau trên UMAP có gene expression thực sự tương tự — là đáng tin cậy.
2.4. Nghiên Cứu Nền Tảng: Hệ Sinh Thái Melanoma
Tirosh et al. (2016) công bố trên Science phân tích scRNA-seq đầu tiên trên khối u melanoma di căn ở người, với 4.645 tế bào từ 19 bệnh nhân. Kết quả tiết lộ điều mà RNA-seq bulk không thể thấy: trong cùng một khối u, tế bào melanoma không đồng nhất mà tồn tại ở nhiều trạng thái phân tử (molecular states) khác nhau — một số biểu hiện chương trình tăng sinh nhanh, một số biểu hiện chương trình xâm lấn, và một số ở trạng thái trung gian chuyển đổi giữa hai cực đó. Nghiên cứu cũng lần đầu vẽ được bản đồ tế bào miễn dịch trong khối u ở độ phân giải đơn tế bào: quần thể tế bào T kiệt sức, tế bào T điều hòa, tế bào NK, và đại thực bào được phân biệt và định lượng chính xác trong cùng một mẫu biopsy. Đây là nền tảng cho toàn bộ lĩnh vực nghiên cứu TME bằng single-cell.
3. scATAC-seq
3.1. Lớp Điều Hòa Epigenome
Gene expression không chỉ phụ thuộc vào chuỗi DNA — nó còn phụ thuộc vào trạng thái đóng/mở của chromatin (chromatin accessibility: mức độ mà các vùng DNA được bao bọc quanh histone có thể tiếp cận bởi các nhân tố phiên mã và enzyme điều hòa). Vùng chromatin mở là vùng đang “hoạt động” — promoter của gene đang được bật, hay enhancer (trình tự tăng cường: vùng DNA không mã hóa có thể kích hoạt phiên mã của gene đích từ khoảng cách hàng trăm nghìn cặp base qua tương tác 3D chromatin) đang gửi tín hiệu tới gene.
Nếu scRNA-seq đo kết quả của điều hòa gene expression (mức RNA), thì scATAC-seq (single-cell Assay for Transposase-Accessible Chromatin by sequencing: đo mức độ tiếp cận chromatin ở độ phân giải đơn tế bào bằng cách dùng enzyme Tn5 transposase để cắt và gắn tag giải trình tự vào các vùng chromatin mở) đo cơ chế điều hòa ở cấp độ epigenome.
3.2. Nguyên Lý Tn5 Transposase
Nguyên lý của scATAC-seq thanh lịch: enzyme Tn5 transposase được nạp trước adapter giải trình tự sẽ ưu tiên chèn vào các vùng DNA không bị bao bọc bởi histone (vùng chromatin mở). Ở các vùng bị cuộn chặt quanh nucleosome, Tn5 không thể tiếp cận. Sau khi cắt và gắn adapter, các đoạn DNA từ vùng mở được khuếch đại và giải trình tự. Kết quả là một peak profile (bản đồ đỉnh tiếp cận chromatin) cho từng tế bào — cho biết vùng regulatory nào đang “mở” trong tế bào đó.
Buenrostro et al. (2015) lần đầu thực hiện scATAC-seq trên 254 tế bào, chứng minh rằng mỗi loại tế bào máu có cảnh quan regulatory (regulatory landscape) đặc trưng — bộ enhancer và promoter mở khác nhau — phản ánh bản sắc phân tử của tế bào ở lớp epigenome. Kể từ đó, scATAC-seq đã được dùng rộng rãi để lập bản đồ cấu trúc regulatory của phát triển, bệnh lý, và đáp ứng điều trị.
3.3. Sức Mạnh Bổ Trợ với scRNA-seq
Dữ liệu scATAC-seq tích hợp với scRNA-seq giúp trả lời một câu hỏi sâu hơn: trong một tế bào cụ thể, vùng regulatory nào đang mở và dẫn đến biểu hiện gene downstream nào? Sự tương quan này cho phép suy ra cis-regulatory network (mạng lưới điều hòa cis: mạng lưới kết nối enhancer với gene đích mà nó điều hòa trong không gian 3D nhiễm sắc thể của từng tế bào). Đây là thông tin cơ chế mà không thể suy ra từ gene expression đơn thuần.
4. Proteomics Đơn Tế Bào
4.1. CyTOF
Trước khi các công nghệ RNA-based chiếm ưu thế, CyTOF (Cytometry by Time-Of-Flight: kỹ thuật đo khối lượng ion của các isotope kim loại nặng gắn trên kháng thể thay vì đo fluorescence như flow cytometry truyền thống, cho phép đo đồng thời 40–60 protein trên mỗi tế bào mà không bị giới hạn bởi sự chồng lấp phổ của dye huỳnh quang) đã mang lại khả năng phân tích protein đơn tế bào ở quy mô chưa từng có.
Flow cytometry cổ điển bị giới hạn bởi sự chồng lấp phổ của fluorescence — thực tế thường không quá 10–15 marker cùng lúc. CyTOF thay thế dye huỳnh quang bằng isotope kim loại (metal isotopes: các đồng vị kim loại nặng không tồn tại tự nhiên trong tế bào sinh học, gắn lên kháng thể và phát hiện bằng khối phổ theo khối lượng nguyên tử) với dải khối lượng nguyên tử không chồng chéo nhau. Nhờ vậy, CyTOF đo được 40–60 protein trong một lần chạy.
Nghiên cứu nền tảng của Bendall et al. (2011) trên Science dùng CyTOF với 34 marker để phân tích quỹ đạo biệt hóa tế bào B và T trong máu người, lần đầu vạch ra differentiation trajectory (quỹ đạo biệt hóa: dòng chảy liên tục qua các trạng thái phân tử trung gian từ tế bào tiền thân đến tế bào trưởng thành) ở độ phân giải đơn tế bào với thông tin protein thực sự.
4.2. CITE-seq
CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by sequencing: kỹ thuật đo đồng thời RNA và protein bề mặt tế bào trong cùng một tế bào bằng cách gắn kháng thể với chuỗi DNA oligonucleotide duy nhất — Antibody-Derived Tag, ADT — và giải trình tự ADT cùng với cDNA) là bước hợp nhất quan trọng nhất trong early multi-modal single-cell.
Nguyên lý: kháng thể chống protein bề mặt tế bào (ví dụ CD3, CD8, CD19) được gắn với một chuỗi DNA barcode duy nhất. Khi tế bào được ủ với cocktail kháng thể rồi đưa vào hệ thống droplet, mỗi droplet bẫy một tế bào cùng tất cả kháng thể gắn trên bề mặt. Sau giải trình tự, mỗi tế bào cung cấp cả RNA profile (gene expression toàn genome) lẫn protein profile (lượng protein bề mặt tế bào định lượng qua ADT count). Stoeckius et al. (2017) công bố CITE-seq với 13 protein marker cùng toàn bộ transcriptome, chứng minh rằng protein và RNA thường bổ sung thông tin cho nhau — đặc biệt quan trọng trong miễn dịch học vì nhiều marker phân loại tế bào là protein bề mặt (CD4, CD8, CD19…) mà RNA không đo chính xác do không tương quan hoàn toàn với protein.
5. Spatial Transcriptomics
5.1. Vấn Đề Của Vị Trí Không Gian
scRNA-seq và các công nghệ đơn tế bào dạng suspension đòi hỏi phá vỡ cấu trúc mô (tissue dissociation: tách mô thành dung dịch tế bào đơn lẻ) trước khi phân tích. Điều này loại bỏ hoàn toàn thông tin về vị trí không gian (spatial context: tọa độ của mỗi tế bào trong mô, tương tác vật lý với tế bào lân cận, và cấu trúc kiến trúc đặc trưng của mô). Tuy nhiên, vị trí tế bào trong mô không phải là ngẫu nhiên — nó quyết định tín hiệu tế bào đó nhận từ môi trường, và thường liên quan trực tiếp đến chức năng.
Spatial transcriptomics (transcriptomics không gian: nhóm công nghệ đo lường gene expression đồng thời với việc bảo tồn thông tin vị trí không gian của từng điểm đo trong mô) giải quyết giới hạn này và được Nature Methods bình chọn là Method of the Year 2020.
5.2. Array-Based: Visium
Visium (10x Genomics) — thương mại hóa từ nền tảng gốc được Ståhl et al. (2016) công bố trên Science — dùng một capture array (mảng bắt giữ: tấm kính phủ mạng lưới đều đặn các điểm tròn nhỏ, mỗi điểm chứa hàng nghìn oligonucleotide poly-dT với spatial barcode duy nhất mã hóa tọa độ x–y). Mô cắt lát mỏng đặt lên tấm kính, RNA khuếch tán từ tế bào được bắt bởi oligonucleotide tại điểm gần nhất. Sau giải trình tự, spatial barcode xác định tọa độ nguồn gốc của từng read trên tấm kính — và vì biết tọa độ tấm kính so với mô, ta biết RNA đó đến từ vùng mô nào.
Mỗi spot của Visium có đường kính 55 micromet và thường chứa 2–10 tế bào — không phải đơn tế bào thực sự mà là độ phân giải dưới mô (sub-cellular resolution). Visium HD thế hệ mới giảm đường kính spot xuống còn 2 micromet, tiếp cận độ phân giải đơn tế bào thực sự.
5.3. Imaging-Based: MERFISH và seqFISH+
Một hướng kỹ thuật hoàn toàn khác là dùng in situ hybridization (lai ngay trong mô: phát hiện RNA trực tiếp trong lát mô nguyên vẹn mà không cần giải trình tự off-slide) kết hợp với imaging nhiều vòng để “đọc” danh tính của từng phân tử RNA riêng lẻ.
MERFISH (Multiplexed Error-Robust Fluorescence In Situ Hybridization: kỹ thuật dùng nhiều vòng lai huỳnh quang với mã hóa barcode nhị phân để xác định danh tính từng phân tử RNA trong mô nguyên vẹn) và seqFISH+ dùng nguyên lý tương tự: mỗi gene được gán một mã barcode nhị phân duy nhất qua nhiều vòng imaging. Trong mỗi vòng, probe huỳnh quang phát sáng hoặc không tùy vị trí trong mã — sau nhiều vòng, chuỗi tín hiệu xác nhận danh tính gene. Kết quả là hình ảnh của mô, trong đó mỗi điểm sáng là một phân tử RNA riêng lẻ với danh tính đã biết.
Hai công nghệ này đạt độ phân giải phân tử đơn — xác định từng phân tử RNA trong không gian 3D của mô — và bảo tồn hoàn toàn kiến trúc mô. Nhược điểm: chỉ đọc được subset gene thiết kế sẵn (thường 300–5.000 gene) thay vì toàn bộ transcriptome như Visium, và quy trình imaging nhiều vòng rất tốn thời gian.
(xem Hình 3.1 cho so sánh hai hướng spatial transcriptomics)
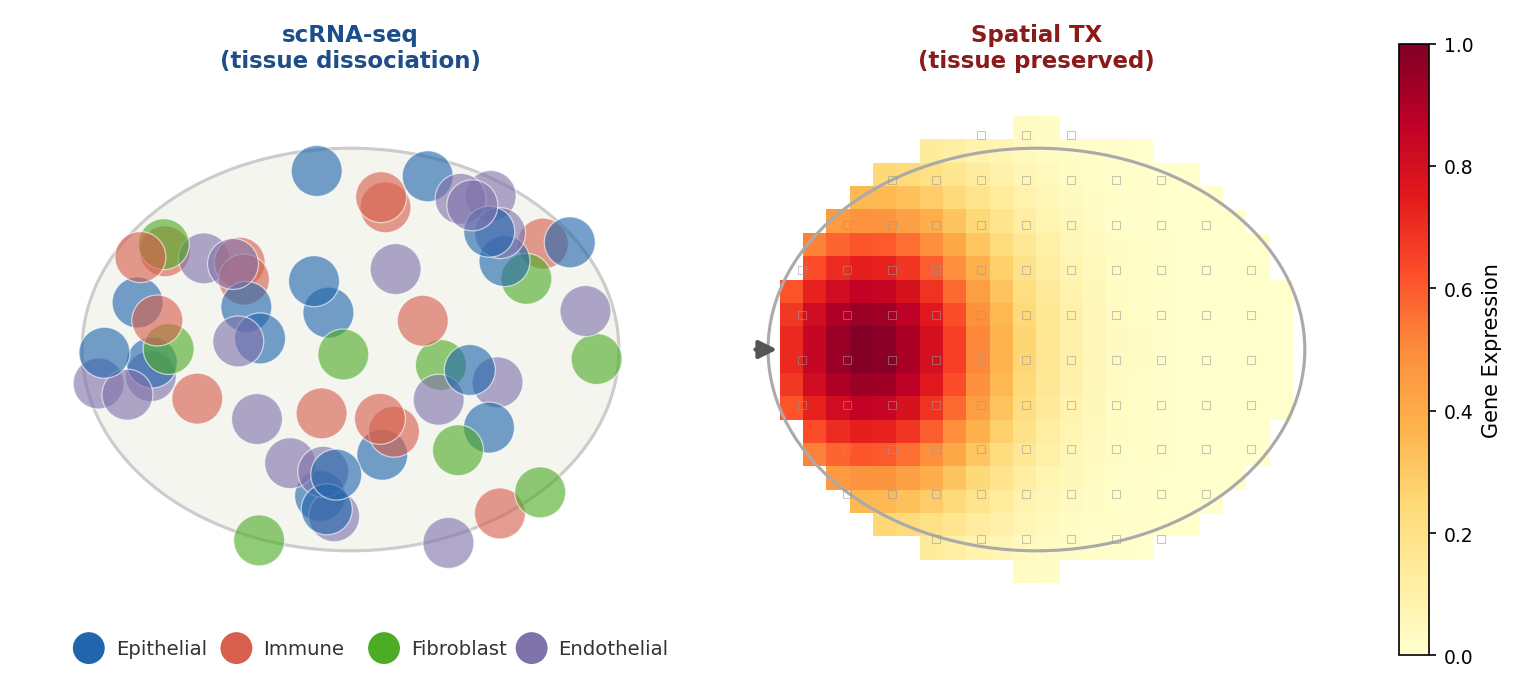 Hình 3.1. Hai hướng tiếp cận spatial transcriptomics: array-based (Visium — RNA khuếch tán và bị bắt bởi barcode trên capture array) và imaging-based (MERFISH/seqFISH — probe huỳnh quang phát hiện từng phân tử RNA trực tiếp trong mô nguyên vẹn).
Hình 3.1. Hai hướng tiếp cận spatial transcriptomics: array-based (Visium — RNA khuếch tán và bị bắt bởi barcode trên capture array) và imaging-based (MERFISH/seqFISH — probe huỳnh quang phát hiện từng phân tử RNA trực tiếp trong mô nguyên vẹn).
6. Multi-Modal Single-Cell Omics
6.1. Tại Sao Đo Nhiều Lớp Cùng Lúc
Mỗi công nghệ đơn tế bào đo một lớp thông tin: scRNA-seq đo transcriptome, scATAC-seq đo chromatin accessibility, CITE-seq thêm protein. Nhưng câu hỏi sinh học thực sự thú vị không phải là “RNA nói gì?” hay “Chromatin nói gì?” mà là “Trong cùng một tế bào, mối quan hệ nhân quả giữa chromatin mở, gene expression, và protein bề mặt là gì?” Từng câu hỏi đơn lẻ có thể trả lời bằng từng công nghệ riêng, nhưng câu hỏi nhân quả đòi hỏi đo lường paired (cùng tế bào, nhiều lớp đồng thời).
6.2. Các Nền Tảng Multi-Modal
10x Multiome cho phép đo đồng thời RNA và ATAC (chromatin accessibility) từ cùng một tế bào, dùng cùng droplet barcoding. Điều này tạo ra dữ liệu truly paired — cùng một tế bào có cả gene expression profile lẫn chromatin accessibility profile — thay vì phải suy luận bằng tích hợp thống kê từ hai thực nghiệm khác nhau trên các tế bào khác nhau.
TEA-seq và DOGMA-seq đẩy xa hơn: đo RNA, ATAC, và protein surface marker từ cùng một tế bào trong ba lớp đồng thời. Những công nghệ này đặt nền móng cho hiểu biết về cảnh quan phân tử đa chiều (multi-dimensional molecular landscape) của từng tế bào — từ epigenome qua transcriptome đến proteome — trong một thực nghiệm duy nhất.
6.3. Thách Thức Tích Hợp Dữ Liệu
Multi-modal single-cell omics tạo ra thách thức phân tích mới: làm thế nào để tích hợp hai hay nhiều ma trận dữ liệu có kích thước và đặc tính thống kê hoàn toàn khác nhau — RNA counts vs binary ATAC peaks vs protein counts — vào một không gian biểu diễn chung? Công cụ như Seurat WNN (Weighted Nearest Neighbor: trọng bình quân láng giềng gần nhất trong không gian đa phương thức), MOFA+ (Multi-Omics Factor Analysis), và scVI (variational inference cho single-cell) dùng các chiến lược học máy và mô hình sinh để học không gian tích hợp từ tất cả các lớp. Chúng tôi sẽ đề cập chi tiết các pipeline phân tích trong bài tutorial riêng.
(xem Hình 4.1 cho so sánh công nghệ)
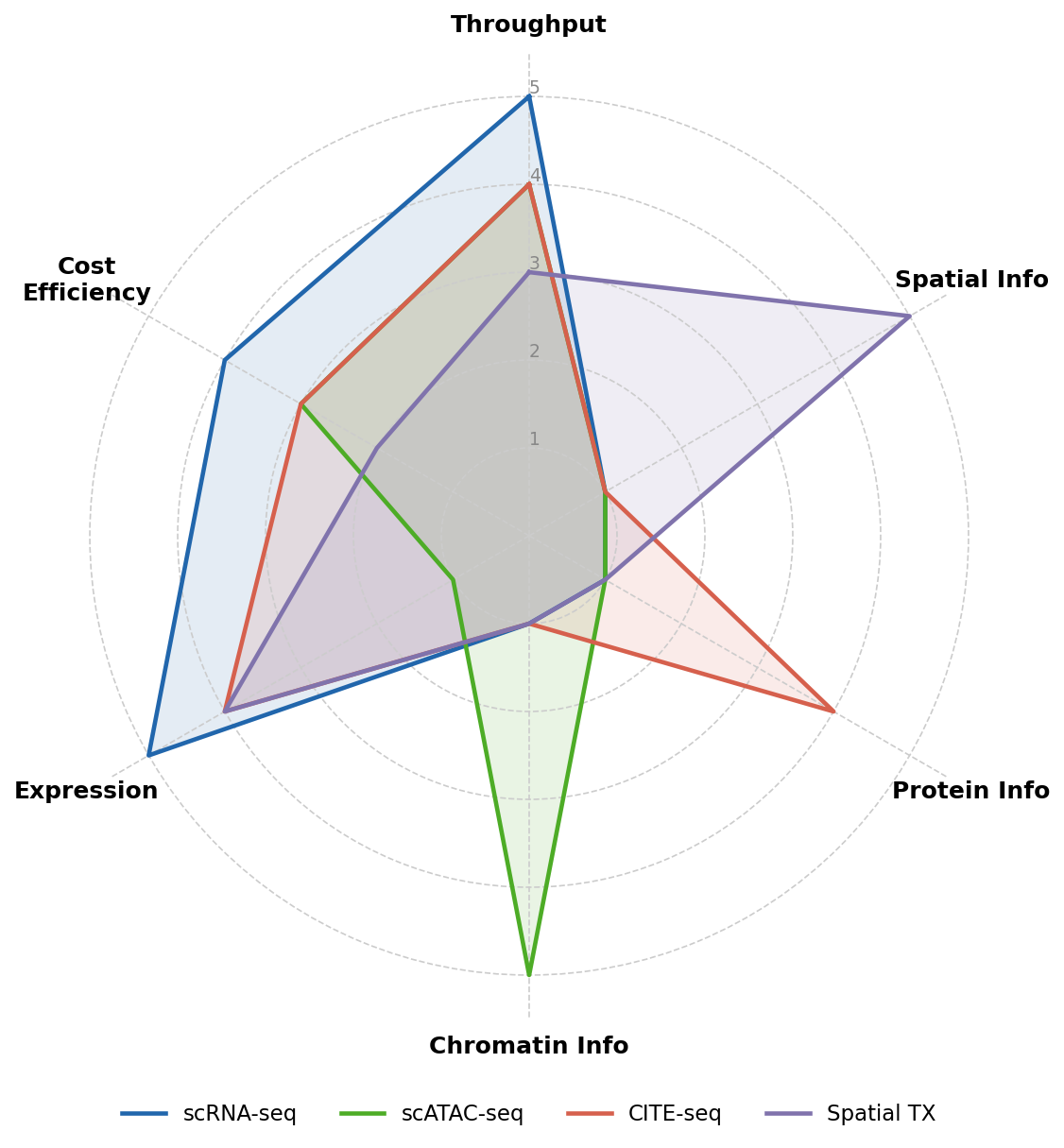 Hình 4.1. Radar chart so sánh bốn công nghệ single-cell omics chính trên sáu tiêu chí: throughput, thông tin không gian, thông tin protein, thông tin chromatin, chi phí hiệu quả, và độ phân giải đơn tế bào.
Hình 4.1. Radar chart so sánh bốn công nghệ single-cell omics chính trên sáu tiêu chí: throughput, thông tin không gian, thông tin protein, thông tin chromatin, chi phí hiệu quả, và độ phân giải đơn tế bào.
7. Các Nghiên Cứu Nền Tảng
7.1. Human Cell Atlas
Human Cell Atlas (HCA: Bản đồ tế bào người — dự án quốc tế nhằm xác định và lập bản đồ phân tử của toàn bộ các loại tế bào trong cơ thể người khỏe mạnh bằng các công nghệ single-cell omics, lấy cảm hứng từ Human Genome Project nhưng hướng tới bản đồ tế bào thay vì bản đồ trình tự DNA) là một trong những dự án khoa học tham vọng nhất thập kỷ này. Được Regev và Teichmann khởi xướng năm 2017, HCA đặt mục tiêu vẽ bản đồ gene expression profile của từng loại tế bào trong mọi mô và cơ quan của cơ thể người — tạo ra một atlas tham chiếu (reference atlas: bản đồ phân loại tế bào chuẩn để so sánh với các trạng thái bệnh lý, phát triển, và can thiệp điều trị) cho sinh học phân tử thế kỷ 21.
Các công bố HCA đã mang lại nhiều khám phá bất ngờ: trong ruột non người, xác định được hơn 10 kiểu tế bào biểu mô chưa từng được mô tả đầy đủ. Trong phổi, atlas single-cell tiết lộ tế bào AT0 (airway transit-amplifying cells: tế bào chuyển tiếp giữa tế bào gốc và tế bào biệt hóa trong biểu mô đường thở) chưa từng được biết đến. Tabula Sapiens (The Tabula Sapiens Consortium, 2022) là dự án đồng hành, xây dựng atlas của 33 mô người từ các donor cùng cá thể, cho phép phân biệt biến thể giữa mô với biến thể giữa người.
7.2. Human Brain Cell Atlas
Não người là cơ quan phức tạp nhất được biết đến, với ước tính 170 tỷ tế bào thuộc hàng trăm loại. Trước khi single-cell omics ra đời, phân loại tế bào thần kinh chủ yếu dựa trên hình thái và điện sinh lý — hai đặc tính đòi hỏi chuyên gia và không thể áp dụng ở quy mô lớn.
Allen Brain Cell Atlas và các nghiên cứu đồng hành đã phân tích hàng triệu tế bào não người bằng scRNA-seq, phát hiện hơn 3.000 loại tế bào — phần lớn chưa có tên trong y văn. Đặc biệt, các loại inhibitory neurons (tế bào thần kinh ức chế biểu hiện GABA) phong phú và đa dạng hơn nhiều so với ước lượng trước, với nhiều loại có phân bố khu vực não rất đặc hiệu. Đây là ví dụ điển hình về cách single-cell omics buộc phải cập nhật lại toàn bộ bảng phân loại của một lĩnh vực y sinh học đã có lịch sử hàng trăm năm.
Spatial transcriptomics đóng vai trò bù trừ: bằng cách ánh xạ các loại tế bào từ scRNA-seq lên dữ liệu spatial Visium của lát não, nhà nghiên cứu vừa biết “loại tế bào này là gì” (từ scRNA-seq) vừa biết “loại tế bào này ở đâu trong não” (từ spatial) — thông tin mà không công nghệ đơn lẻ nào cung cấp được.
7.3. COVID-19 Cell Atlas
Đại dịch COVID-19 tạo ra một tình huống nghiên cứu đặc biệt: cần hiểu nhanh cơ chế bệnh sinh của một tác nhân hoàn toàn mới. COVID-19 Cell Atlas tập hợp dữ liệu scRNA-seq từ hàng chục nhóm nghiên cứu toàn cầu, phân tích bạch cầu máu ngoại vi và mô phổi của bệnh nhân ở các mức độ nặng khác nhau.
Kết quả cho thấy cơn bão cytokine (cytokine storm: trạng thái kích hoạt hệ miễn dịch quá mức gây tổn thương mô và suy đa tạng) không phải là một hiện tượng đồng nhất — nó là kết quả của một loạt đại thực bào bị kích hoạt theo trạng thái profibrotic (gây xơ hóa) cùng với sự suy giảm đồng thời của tế bào T chức năng. Dữ liệu single-cell cũng tiết lộ rằng ACE2 — thụ thể mà SARS-CoV-2 dùng để xâm nhập tế bào — được biểu hiện cao chủ yếu trong một kiểu tế bào biểu mô khoang mũi rất đặc hiệu, thông tin trực tiếp định hướng thiết kế vaccine aerosol và liệu pháp ức chế xâm nhập.
7.4. Cancer Single-Cell Atlases
Từ nghiên cứu tiên phong của Tirosh et al. (2016), nhiều cancer cell atlas chuyên biệt đã được xây dựng: Human Tumor Atlas Network (HTAN) tập hợp single-cell data từ nhiều loại ung thư với tiêu chuẩn dữ liệu thống nhất. Pan-Cancer T-Cell Atlas lập bản đồ cách tế bào T thay đổi chức năng trong 21 loại ung thư khác nhau, xác định các trạng thái tế bào T kiệt sức có khả năng đáp ứng với liệu pháp checkpoint inhibitor (thuốc ức chế điểm kiểm soát miễn dịch: nhóm thuốc giải phóng tế bào T khỏi trạng thái kiệt sức do khối u áp đặt, cho phép tế bào T tấn công ung thư trở lại) — thông tin trực tiếp hướng dẫn phát triển biomarker phân tầng bệnh nhân.
8. So Sánh Và Hướng Dẫn Chọn Công Nghệ
Không có công nghệ single-cell omics nào phù hợp với mọi câu hỏi nghiên cứu. Bảng dưới đây tóm tắt điểm mạnh, điểm yếu, và tình huống sử dụng điển hình của từng nền tảng:
| Công nghệ | Thông tin đo | Throughput | Spatial | Use Case Điển Hình |
|---|---|---|---|---|
| scRNA-seq (10x) | Transcriptome | Rất cao | Không | Phân loại kiểu tế bào, TME |
| Smart-seq2 | Transcriptome full-length | Thấp | Không | Tế bào hiếm, isoform analysis |
| scATAC-seq | Chromatin accessibility | Cao | Không | Regulatory elements, lineage |
| CITE-seq | RNA + protein surface | Cao | Không | Miễn dịch học, phenotyping |
| CyTOF | Protein (40–60 markers) | Cao | Không | Immunophenotyping chi tiết |
| Visium | Transcriptome + spatial | Trung bình | Spot (~55 μm) | Mô học phân tử |
| MERFISH/seqFISH+ | RNA in situ (subset genes) | Thấp–trung | Phân tử đơn | Não, kiến trúc mô |
| 10x Multiome | RNA + ATAC | Cao | Không | Regulatory + expression |
Bảng 8.1. So sánh các nền tảng single-cell omics chính về thông tin đo lường, throughput, khả năng spatial, và use case điển hình.
Một số nguyên tắc thực tiễn khi chọn công nghệ:
- Mục tiêu là phân loại tế bào và hiểu cấu trúc quần thể: scRNA-seq 10x là điểm bắt đầu tốt nhất — throughput cao, chi phí hợp lý, pipeline phân tích trưởng thành nhất.
- Câu hỏi liên quan đến điều hòa gene và cơ chế epigenomic: kết hợp scATAC-seq hoặc 10x Multiome với scRNA-seq.
- Nghiên cứu miễn dịch học cần phân loại protein chính xác: CITE-seq hoặc CyTOF cung cấp thông tin protein mà RNA không thể thay thế.
- Kiến trúc mô và vị trí tế bào là trung tâm của câu hỏi: Visium cho tổng quan toàn mô, MERFISH/seqFISH+ cho độ phân giải phân tử đơn.
- Cần hiểu nhân quả giữa nhiều lớp phân tử: 10x Multiome hoặc CITE-seq cung cấp dữ liệu paired thực sự, tránh sai số từ tích hợp thống kê cross-modality.
Kết Luận
Single-cell omics đã biến đổi sinh học từ khoa học đo “trung bình” thành khoa học đo “từng cá thể tế bào”. Không đầy một thập kỷ từ 2015 đến nay, lĩnh vực này đã vẽ xong phần lớn bản đồ kiểu tế bào của cơ thể người, giải mã hệ sinh thái của hàng chục loại ung thư, cung cấp cái nhìn cơ chế phân tử chi tiết chưa từng có vào bệnh nhiễm trùng và bệnh thần kinh, và buộc chúng ta phải viết lại nhiều phần của sinh học phân tử truyền thống.
Điều đáng chú ý hơn là các công nghệ này không dừng lại: độ phân giải tăng (Visium HD, MERFISH 10.000 gene), chi phí giảm, và tích hợp đa lớp multi-modal ngày càng trở nên khả thi. Bước tiếp theo của lĩnh vực là single-cell multi-omics tích hợp toàn diện kết hợp với spatial resolution — đo đồng thời genome, epigenome, transcriptome, proteome trong không gian mô 3D. Những sinh viên và nhà nghiên cứu tham gia lĩnh vực này hôm nay đang đứng ở đầu một trong những cơ hội khám phá rộng lớn nhất trong lịch sử sinh học hiện đại.
Tài Liệu Tham Khảo
Buenrostro, J. D., Wu, B., Litzenburger, U. M., Ruff, D., Gonzales, M. L., Snyder, M. P., Chang, H. Y., & Greenleaf, W. J. (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 523(7561), 486–491. https://doi.org/10.1038/nature14590
Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., Trombetta, J. J., Weitz, D. A., Sanes, J. R., Shalek, A. K., Regev, A., & McCarroll, S. A. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Regev, A., Teichmann, S. A., Lander, E. S., Bhatt, D., Bhattacharya, J., Bhavsar, P., Butler, A., & the HCA Working Group. (2017). The Human Cell Atlas. eLife, 6, e27041. https://doi.org/10.7554/eLife.27041
Ståhl, P. L., Salmén, F., Vickovic, S., Lundmark, A., Navarro, J. F., Magnusson, J., Giacomello, S., Asp, M., Westholm, J. O., Huss, M., Mollbrink, A., Linnarsson, S., Codeluppi, S., Borg, Å., Pontén, F., Costea, P. I., Sahlén, P., Mulder, J., Bergmann, O., & Frisén, J. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 353(6294), 78–82. https://doi.org/10.1126/science.aaf2403
Stoeckius, M., Hafemeister, C., Stephenson, W., Houck-Loomis, B., Chattopadhyay, P. K., Swaminathan, H., Satija, R., & Smibert, P. (2017). Simultaneous epitope and transcriptome measurement in single cells. Nature Methods, 14(9), 865–868. https://doi.org/10.1038/nmeth.4380
The Tabula Sapiens Consortium. (2022). The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science, 376(6588), eabl4896. https://doi.org/10.1126/science.abl4896
Tirosh, I., Izar, B., Prakadan, S. M., Wadsworth, M. H., Treacy, D., Trombetta, J. J., Rotem, A., Rodman, C., Lian, C., Murphy, G., Fallahi-Sichani, M., Dutton-Regester, K., Lin, J.-R., Cohen, O., Shah, P., Lu, D., Genshaft, A. S., Hughes, T. K., Ziegler, C. G. K., & Garraway, L. A. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science, 352(6282), 189–196. https://doi.org/10.1126/science.aad0501
Zheng, G. X., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., Gregory, M. T., Shuga, J., Montesclaros, L., Underwood, J. G., Masquelier, D. A., Nishimura, S. Y., Schnall-Levin, M., Wyatt, P. W., Hindson, C. M., & Bielas, J. H. (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications, 8, 14049. https://doi.org/10.1038/ncomms14049
Explore more like this
Gene Regulatory Network: Từ Phân Tử Đến Multi-Omics Và Ứng Dụng Lâm Sàng
Genome của người chứa khoảng 20.000 gene mã hóa protein, nhưng không phải gene nào cũng hoạt động ở mọi tế bào trong mọi thời điểm. Một tế bào gan,...
Từ Dữ Liệu Thô Đến Biến Thể Di Truyền: Genome Alignment, Variant Calling, QC và Annotation
Khi một mẫu DNA được đưa vào máy giải trình tự, kết quả trả về không phải là một câu trả lời hoàn chỉnh về bộ genome của cá thể...
Transcriptomics: Hành Trình Giải Mã Hệ Phiên Mã và Các Công Nghệ Chủ Chốt
Tế bào người chứa khoảng 3,2 tỷ cặp bazơ DNA — nhưng không phải tất cả đều được “đọc” cùng một lúc, hay ở mọi loại tế bào. Một tế...
Comments