Từ Dữ Liệu Thô Đến Biến Thể Di Truyền: Genome Alignment, Variant Calling, QC và Annotation

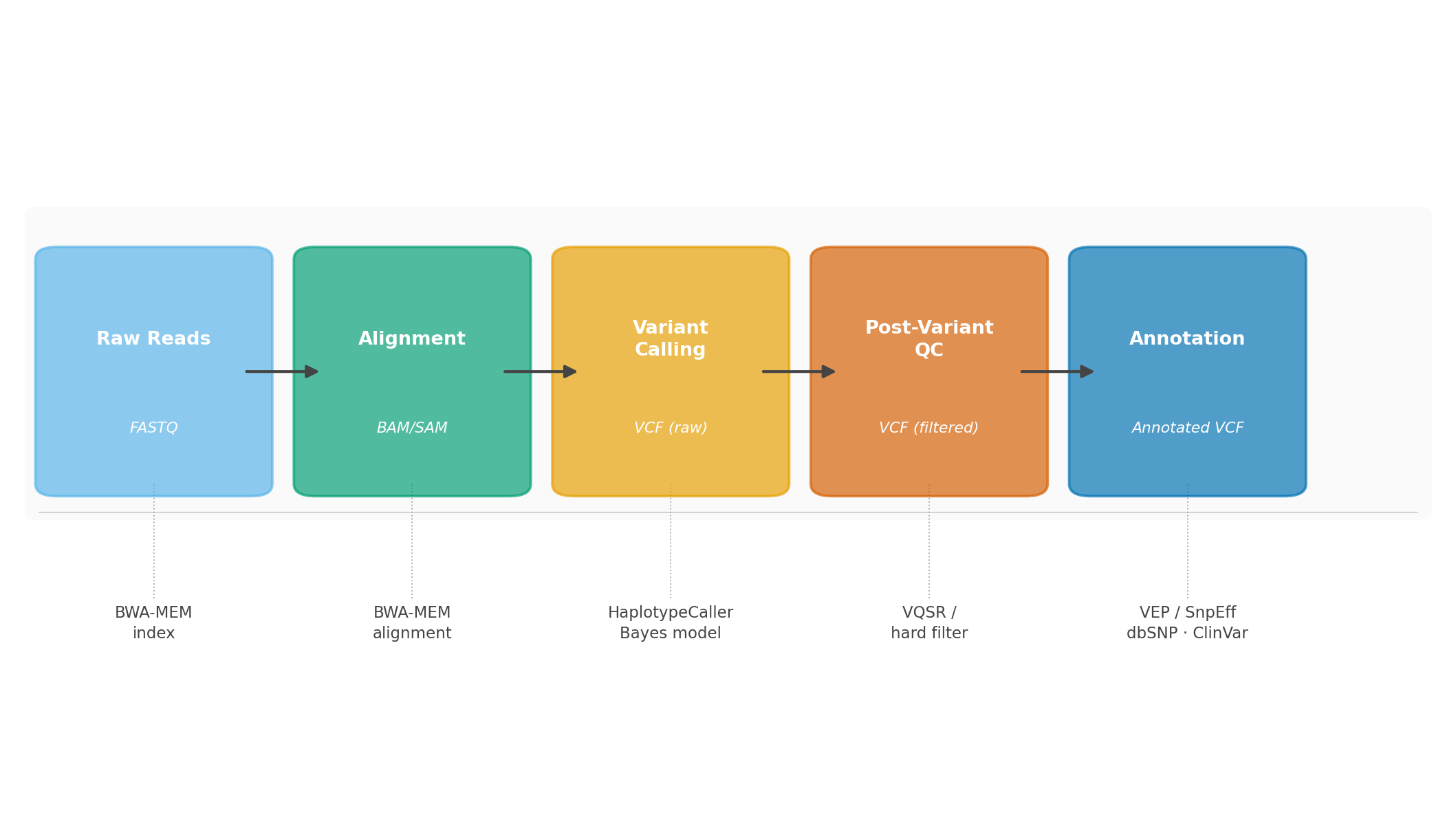
Khi một mẫu DNA được đưa vào máy giải trình tự, kết quả trả về không phải là một câu trả lời hoàn chỉnh về bộ genome của cá thể đó. Với một thí nghiệm WGS (whole genome sequencing — giải trình tự toàn bộ genome) ở độ phủ 30x thông thường, máy sẽ trả về khoảng 900 triệu đoạn ngắn, mỗi đoạn dài 150 ký tự, hoàn toàn rời rạc và không có thứ tự. Tổng thể chúng chứa đủ thông tin để tái dựng lại bộ genome 3,2 tỷ ký tự của cá thể đó — nhưng chỉ khi biết cách “đọc” chúng đúng cách.
Việc đặt từng mảnh nhỏ vào đúng vị trí trên bản đồ genome, nhận diện những điểm khác biệt so với tham chiếu chuẩn, loại bỏ những khác biệt giả tạo do nhiễu kỹ thuật, rồi giải thích ý nghĩa sinh học và lâm sàng của từng biến thể — đó là bốn chương của quy trình phân tích genomics hiện đại. Mỗi chương là một bài toán tính toán riêng biệt với những thách thức đặc thù. Bài viết này lý giải trực giác và cơ chế hoạt động cấp cao của từng bước — không phải từ góc độ công thức toán học hay cú pháp phần mềm, mà từ góc độ tại sao chúng ta làm vậy, thuật toán giải quyết bài toán như thế nào, và điểm khó nằm ở đâu.
1. Alignment
1.1. Bài Toán Cốt Lõi
Read alignment (căn chỉnh dữ liệu giải trình tự) là bước đặt từng đoạn ngắn — gọi là read — vào đúng vị trí của nó trên reference genome. Hãy hình dung bạn xé nát một quyển sách 3 tỷ ký tự thành 900 triệu mảnh giấy nhỏ, mỗi mảnh chứa 150 ký tự, rồi trộn ngẫu nhiên tất cả. Bài toán alignment là: cho mỗi mảnh giấy đó, hãy tìm xem nó xuất phát từ vị trí nào trong quyển sách. Ngay cả với máy tính hiện đại, nếu duyệt từng mảnh một cách ngây thơ — so sánh tuần tự với mọi vị trí trên genome 3,2 tỷ ký tự — tổng số phép tính sẽ là khoảng 900 triệu × 3,2 tỷ = hơn 10^18 phép tính. Với tốc độ 10 tỷ phép/giây của CPU hiện đại, cần hàng trăm triệu giây — tức là hàng chục năm cho một mẫu. Điều này là không chấp nhận được.
Hiểu được giới hạn đó, mọi phần mềm alignment hiện đại đều xây dựng trước một index (chỉ mục) của reference genome — tương tự như mục lục cuối quyển sách hay index của cơ sở dữ liệu — để tra nhanh vị trí gần đúng trước khi khớp chính xác. Việc xây dựng index chỉ cần làm một lần cho mỗi reference genome; sau đó hàng trăm triệu reads có thể tra cứu song song.
1.2. Nền Tảng Khái Niệm: Từ Pairwise Alignment Đến Indexing
Trước khi bàn về index, cần hiểu bài toán alignment đơn giản nhất: so sánh hai chuỗi ký tự để tìm cách căn chỉnh tốt nhất. Pairwise alignment (căn chỉnh cặp đôi) là nền tảng của toàn bộ lĩnh vực này.
Hai thuật toán nền tảng là Needleman-Wunsch (so sánh toàn bộ hai chuỗi — global alignment) và Smith-Waterman (tìm vùng khớp tốt nhất bất kỳ — local alignment). Cả hai đều dùng dynamic programming (lập trình động): điền dần một bảng 2D, mỗi ô đại diện cho điểm alignment tốt nhất giữa một tiền tố của chuỗi này và một tiền tố của chuỗi kia. Khi điền đến ô (i,j), chỉ cần nhìn vào ba ô lân cận — bên trên (deletion), bên trái (insertion), hoặc chéo (match/mismatch) — và chọn cái nào cho điểm cao nhất. Cuối cùng, truy vết ngược lại từ ô có điểm cao nhất để thu được alignment.
Hai thuật toán này cho kết quả chính xác nhất có thể nhưng có độ phức tạp O(n×m) với n, m là độ dài hai chuỗi. Với một read 150bp và genome 3,2 tỷ bp: quá chậm để chạy cho từng read. Và đây chính xác là lý do tại sao indexing ra đời — để tìm nhanh vị trí gần đúng nơi read có thể khớp, sau đó mới dùng Smith-Waterman để căn chỉnh chính xác ở vùng nhỏ đó (thường chỉ vài trăm bp).
1.3. BWT Index: Cỗ Máy Tìm Kiếm Hiệu Quả
Phương pháp indexing chiếm ưu thế tuyệt đối hiện nay là BWT-based (dựa trên Burrows-Wheeler Transform). Trực giác của BWT khá thú vị.
Bắt đầu với chuỗi genome G. Hãy tạo ra tất cả mọi rotation (xoay vòng) của chuỗi đó — nghĩa là lấy đoạn từ vị trí i đến cuối, nối với đoạn từ đầu đến vị trí i−1 — rồi sắp xếp tất cả các rotation đó theo thứ tự bảng chữ cái. Cột đầu tiên của bảng (F) là các ký tự đầu tiên của từng rotation đã sắp xếp; cột cuối (L) là BWT — các ký tự cuối cùng. Nghe có vẻ vô nghĩa, nhưng BWT có một tính chất đặc biệt: các ký tự đứng trước cùng một context sẽ được nhóm lại trong cột L. Ví dụ, tất cả các ký tự đứng trước “GATC” sẽ nằm gần nhau trong L.
Tính chất này, kết hợp với cấu trúc dữ liệu FM-index (Full-text index in Minute space) và suffix array, cho phép tìm kiếm một chuỗi truy vấn bằng cách thu hẹp dần phạm vi tìm kiếm từ cuối chuỗi về đầu — mỗi bước chỉ cần vài thao tác đơn giản. Thay vì so sánh tuyến tính, ta thực hiện một kiểu binary search trên cấu trúc dữ liệu đã được tối ưu. Kết quả: tìm vị trí của một chuỗi 150 ký tự trong genome 3,2 tỷ ký tự chỉ mất vài micro-giây. Và quan trọng hơn, toàn bộ index chỉ cần ~2–3 GB RAM — phù hợp với máy chủ thông thường.
Các công cụ BWA-MEM và Bowtie2 đều xây dựng trên nền tảng FM-index này. BWA-MEM hiện là chuẩn vàng cho WGS và WES người.
1.4. Seed-and-Extend: Cơ Chế Hoạt Động Của BWA-MEM
BWA-MEM không đơn giản chỉ “tra index rồi xong”. Quy trình của nó gồm ba pha khái niệm:
Pha 1 — Seeding (tìm hạt giống): Với mỗi read, BWA-MEM dùng FM-index để tìm các SMEM (Super Maximal Exact Match — đoạn khớp chính xác dài nhất không thể kéo dài thêm mà không mất vị trí khớp). Đây là những đoạn con của read khớp hoàn hảo (100% đồng nhất) với ít nhất một vị trí trên reference. Mỗi SMEM cho biết: “đoạn này của read, tôi biết chắc nó nằm ở đâu, hoặc ở rất ít vị trí trên reference”. Đây là các “neo” định vị ban đầu.
Pha 2 — Chaining (xâu chuỗi): Một read thường cho ra nhiều SMEMs. Nếu nhiều SMEMs trên cùng một read đều trỏ đến cùng một vùng trên reference — với khoảng cách tương đối nhất quán — chúng được xâu thành một chain (chuỗi). Chain là ứng viên cho một alignment. Bước này loại bỏ hầu hết các vị trí không khả thi mà không cần tốn công căn chỉnh chi tiết.
Pha 3 — Extension (mở rộng): Với mỗi chain ứng viên tốt nhất, BWA-MEM chạy banded Smith-Waterman (Smith-Waterman trên dải hẹp) để căn chỉnh chính xác toàn bộ read vào vùng reference đó, bao gồm các mismatches và indels. “Banded” nghĩa là chỉ điền một dải hẹp của bảng DP thay vì toàn bộ — điều này hợp lý vì ta đã biết gần đúng vị trí từ chain, nên alignment chính xác sẽ không lệch quá xa. Kết quả là phần lớn thời gian tính toán được tiết kiệm.
Sức mạnh của cách tiếp cận này: ~95% reads được đặt chính xác chỉ từ bước seeding; bước extension chỉ cần xử lý phần biên và giải quyết những trường hợp phức tạp hơn.
1.5. Vùng Lặp Lại Và Mapping Quality
Khoảng 45% genome người là repetitive regions (vùng trình tự lặp lại) — những đoạn trình tự gần giống nhau xuất hiện ở hàng nghìn vị trí trên genome, như các phần tử LINE, SINE, và satellite repeats. Khi một read xuất phát từ vùng lặp, FM-index sẽ tìm thấy hàng nghìn vị trí khớp ngang nhau — thuật toán không thể xác định vị trí thật của read.
Đây là lý do alignment không chỉ trả về vị trí mà còn trả về MAPQ (mapping quality — điểm chất lượng căn chỉnh). MAPQ được tính theo thang Phred: MAPQ = 60 có nghĩa là xác suất đặt nhầm chỉ là 1 trong 1 triệu; MAPQ = 20 là 1%; MAPQ = 0 nghĩa là read đặt tốt nhất chỉ ngang bằng với vị trí tốt thứ nhì — việc chọn vị trí nào cũng như nhau, không đáng tin. BWA-MEM quy ước: read khớp duy nhất và hoàn hảo nhận MAPQ 60; read đa vị trí (multimapper) nhận MAPQ 0 hoặc rất thấp.
Điểm MAPQ này sẽ được dùng như bộ lọc trong variant calling và QC: biến thể gọi chủ yếu từ các reads có MAPQ thấp rất nhiều khả năng là artifact do alignment sai.
1.6. Gapped Alignment Và Xử Lý Indel
Không phải mọi sự khác biệt giữa read và reference đều là mismatch một ký tự. Indel (insertion và deletion — đoạn chèn thêm hoặc thiếu mất so với reference) là loại biến thể quan trọng thứ hai sau SNV (single nucleotide variant). Để xử lý indels, alignment phải cho phép gaps.
Trong Smith-Waterman banded, mỗi gap có một gap open penalty (phí mở gap) và gap extension penalty (phí kéo dài gap). Gap open penalty cao hơn nhiều so với gap extension — điều này phản ánh thực tế sinh học: trong genome người, indels thường là một đoạn liên tiếp bị mất hoặc thêm vào, không phải nhiều điểm rời rạc. Cấu trúc phí phạt này “hướng dẫn” thuật toán ưu tiên một gap dài hơn nhiều gap ngắn rời rạc.
Kết quả alignment của mỗi read được lưu dưới dạng CIGAR string — một chuỗi ký hiệu súc tích mô tả cấu trúc alignment: ví dụ “50M2I48M” có nghĩa là 50 ký tự khớp, 2 ký tự insertion, 48 ký tự khớp. Toàn bộ file alignment được lưu định dạng BAM (Binary Alignment Map) — định dạng nhị phân nén của SAM (Sequence Alignment Map), sẵn sàng cho bước variant calling.
2. Variant Calling
2.1. Góc Nhìn Pileup
Sau khi alignment hoàn tất, hãy tưởng tượng nhìn từ trên xuống genome: tại mỗi vị trí, nhiều reads chồng lên nhau như một cột gạch. Nhìn vào cột gạch tại từng vị trí — gọi là pileup — cho thấy: hầu hết vị trí mọi reads đều đồng thuận với reference; tại một số vị trí, một phần reads báo cáo một ký tự khác.
Cách tiếp cận ngây thơ nhất: nếu ≥20% reads tại vị trí P báo cáo ký tự B ≠ reference, gọi đây là variant. Phương pháp này được các công cụ sơ khai áp dụng. Vấn đề là nó hoàn toàn bỏ qua: chất lượng của từng nucleotide riêng lẻ, chất lượng alignment của từng read, mối liên hệ giữa các vị trí lân cận, và quan trọng nhất — nó xử lý từng vị trí độc lập với nhau, trong khi thực tế các biến thể di truyền thường được kế thừa cùng nhau trên cùng một nhiễm sắc thể.
2.2. Active Region Detection
GATK HaplotypeCaller — thuật toán variant calling phổ biến nhất trong WGS/WES — không xử lý toàn bộ genome theo kiểu pileup đơn giản. Bước đầu tiên của nó là xác định active region (vùng tích cực): những vùng trên genome thực sự có khả năng chứa biến thể.
Cơ chế phát hiện active region: quét qua pileup; nếu tại một vùng nhỏ có tỉ lệ reads khác reference vượt một ngưỡng nhất định, hoặc có nhiều reads bị soft-clipped (bị cắt một phần khi alignment — dấu hiệu của indel hay structural variant phức tạp), vùng đó được đánh dấu là active. Bên ngoài active regions, algorithm gọi thẳng reference — không có variant. Bên trong active regions, mới áp dụng quy trình đầy đủ và tốn kém hơn.
Bước lọc sớm này tiết kiệm rất nhiều tài nguyên tính toán và giảm false positive từ những vùng hoàn toàn “sạch”.
2.3. Local Reassembly Bằng De Bruijn Graph
Điểm đáng chú ý nhất của HaplotypeCaller là: thay vì nhìn vào từng cột pileup riêng lẻ, nó tái lắp ráp cục bộ toàn bộ haplotype từ reads trong active region đó. Đây là bước tương tự ở mức nhỏ với de novo assembly, nhưng giới hạn trong một cửa sổ ~300–1000 bp.
Cách tái lắp ráp dùng de Bruijn graph (đồ thị de Bruijn): lấy tất cả reads phủ active region, cắt chúng thành các k-mers (những chuỗi con độ dài k, ví dụ k=25). Mỗi k-mer là một nút trong đồ thị; nếu k-mer “ACGTACGT…” theo sau ngay bởi k-mer bắt đầu bằng 24 ký tự cuối của nó, ta vẽ một mũi tên nối. Kết quả là một đồ thị có hướng mô tả tất cả mọi chuỗi trình tự có thể tạo ra từ tập reads đó.
Điểm kỳ diệu ở đây: nếu tại vị trí 450 của đoạn genome đang xét có một SNV (T→C), sẽ có hai nhánh song song đi qua vị trí đó trong đồ thị — một nhánh là path của T (reference), một nhánh là path của C (alternate). Những nhánh rẽ đôi này, gọi là bubble, là các biến thể ứng viên. Mọi đường đi từ nút đầu đến nút cuối của đồ thị là một haplotype ứng viên; enumerating các đường đi này cho ra tập haplotypes cần đánh giá.
Tại sao không nhìn từng vị trí theo pileup? Vì de Bruijn graph xử lý đồng thời nhiều biến thể gần nhau trong cùng một khung, và đặc biệt xử lý indels tốt hơn nhiều. Một indel 5 bp tạo ra một bubble với chiều dài khác nhau ở hai nhánh — điều này được mô hình hóa tự nhiên trong đồ thị, trong khi pileup-based caller sẽ gặp khó khăn vì indels phá vỡ sự căn chỉnh theo cột.
2.4. PairHMM: Tính Xác Suất Likelihood
Sau khi có tập haplotypes ứng viên từ de Bruijn graph, HaplotypeCaller cần đánh giá: với mỗi haplotype ứng viên, nếu đây là trình tự thật, thì các reads quan sát được có xác suất bao nhiêu?
Bước này dùng pair HMM (Hidden Markov Model áp dụng cho pairwise alignment — một mô hình xác suất biểu diễn ngầm ba trạng thái ẩn: Match, Insertion, Deletion). Điểm mấu chốt là: pair HMM không hỏi “read này khớp hay không khớp với haplotype đó?” — nó hỏi “với mọi cách align read này lên haplotype đó, tổng xác suất của tất cả cách align đó là bao nhiêu?”. Bằng cách cộng tất cả khả năng alignment (kể cả alignment với các indels), pair HMM đưa ra một số duy nhất là likelihood — xác suất quan sát được read đó nếu haplotype đó là đúng.
Điều làm pair HMM mạnh hơn alignment đơn giản là: nó sử dụng trực tiếp base quality score của từng ký tự trong read như xác suất lỗi. Một base quality 30 = xác suất lỗi 0.1%; base quality 10 = xác suất lỗi 10%. Vì vậy, một mismatch tại vị trí có base quality thấp ít ảnh hưởng đến likelihood hơn là một mismatch tại vị trí base quality cao. Điều này quan trọng vì đầu và cuối read thường có base quality thấp hơn giữa read.
2.5. Bayesian Genotyping
Sau khi có likelihoods của tất cả reads cho tất cả haplotypes ứng viên, bước cuối là genotyping: quyết định bộ gen kiểu nào là đúng nhất cho cá thể này.
Với sinh vật lưỡng bội như người, mỗi vị trí có hai allele. Các genotype khả dĩ là: ref/ref (đồng hợp tử reference), ref/alt (dị hợp tử), alt/alt (đồng hợp tử alternate). Sử dụng định lý Bayes: xác suất hậu nghiệm của mỗi genotype tỷ lệ với tích của xác suất tiền nghiệm (prior — dựa trên tần số biến thể trong quần thể từ dbSNP) nhân với likelihood của tất cả reads nếu genotype đó là đúng. Genotype có xác suất hậu nghiệm cao nhất được chọn, và GQ (genotype quality score) đo mức độ tự tin của quyết định đó.
Output là file VCF (Variant Call Format) — một bảng danh sách các biến thể với đầy đủ thông tin genotype và các chỉ số chất lượng.
2.6. Joint Calling Và GVCF Mode
Phân tích nhiều mẫu không bao giờ nên gọi variant độc lập cho từng mẫu rồi ghép lại. Joint calling (gọi biến thể đồng thời) mạnh hơn nhiều vì thông tin từ các mẫu hỗ trợ lẫn nhau.
Cơ chế: HaplotypeCaller chạy ở GVCF mode cho mỗi mẫu — trả về một Genomic VCF chứa bằng chứng ở mọi vị trí genome (không chỉ variant sites), kèm theo điểm tin cậy reference cho các vị trí không có biến thể. Sau đó GenomicsDBImport gộp GVCFs của hàng trăm đến hàng nghìn mẫu vào một cơ sở dữ liệu; GenotypeGVCFs chạy joint genotyping trên toàn bộ cohort.
Lợi ích: một biến thể có bằng chứng yếu trong một mẫu nhưng xuất hiện nhất quán ở 50 mẫu trở nên đáng tin cậy hơn nhiều. Một genotype trông như variant trong một mẫu nhưng được gọi reference trong tất cả mẫu còn lại trông giống artifact hơn. Đây là lý do các cohort nghiên cứu lớn như UK Biobank (500.000 cá thể) cho phép phát hiện các biến thể rất hiếm với tần số 0.001% mà không thể phát hiện được trong một mẫu đơn lẻ.
2.7. Germline Và Somatic: Hai Bài Toán Khác Nhau
Germline variant (biến thể dòng mầm) có trong mọi tế bào cơ thể ngay từ khi thụ tinh; expected allele fraction (tỷ lệ reads mang allele đó) là 50% cho dị hợp tử và 100% cho đồng hợp tử — tỷ lệ rõ ràng, dễ phân biệt với nhiễu kỹ thuật.
Somatic variant (biến thể soma) xuất hiện trong quá trình sống, ở một số tế bào nhất định — đây là cơ sở phân tử của ung thư. Bài toán tính toán khó hơn nhiều:
- Khối u là hỗn hợp: tế bào ung thư + tế bào mô đệm lành tính (stromal); thậm chí trong tế bào ung thư, không phải clone nào cũng có cùng mutation (intratumor heterogeneity)
- Một somatic mutation xuất hiện trong 40% tế bào ung thư từ khối u có tumor purity 60% sẽ chỉ có allele fraction = 0.40 × 0.60 = 24% — không xa với ngưỡng nhiễu kỹ thuật
- Somatic variant cần phân biệt khỏi germline variant của cùng cá thể
Mutect2 giải quyết điều này bằng thiết kế khác HaplotypeCaller: nó coi allele fraction là tham số tự do (không bị ràng buộc ở 50%), dùng mẫu normal (mô lành của cùng bệnh nhân) như đối chứng để trừ đi các germline variant, và sử dụng panel of normals (PoN — tập hợp các mẫu normal từ nhiều cá thể khác) để lọc các artifact kỹ thuật lặp lại có hệ thống.
3. Post-Variant QC
3.1. Nguồn Gốc Của False Positive
Ngay cả sau khi HaplotypeCaller/Mutect2 chạy xong, file VCF vẫn chứa một tỷ lệ không nhỏ false positive — những biến thể không tồn tại trong thực tế. Các nguồn gốc chính:
- PCR error: trong quá trình chuẩn bị thư viện, PCR khuếch đại phân tử DNA có thể đưa vào lỗi ngẫu nhiên; những lỗi này xuất hiện trên nhiều reads nhưng không phải biến thể thật
- Systematic sequencer error: Illumina có các context-specific error (ví dụ GGC trinucleotide thường bị đọc sai với tỷ lệ cao hơn trung bình)
- Oxidative DNA damage: 8-oxoguanine (do oxy hóa trong quá trình chuẩn bị mẫu) gây ra artifact G→T đặc trưng trong library prep
- Misalignment trong vùng lặp: reads từ một vị trí lặp được align sai sang vị trí lặp khác, tạo ra “biến thể” ảo giữa hai vị trí lặp có trình tự hơi khác nhau
- Contamination: nếu mẫu bị nhiễm một lượng nhỏ DNA từ cá thể khác, variant của cá thể nhiễm xuất hiện ở allele fraction thấp — trông giống somatic variant
3.2. Các Chiều Annotation Kỹ Thuật
Mỗi variant trong VCF được báo cáo kèm theo hàng chục INFO field — không phải annotation sinh học mà là chỉ số kỹ thuật đo mức độ tin cậy của quyết định gọi biến thể. Các chiều quan trọng nhất:
- QD (Quality by Depth): điểm variant quality chia cho độ phủ tại vị trí đó. Cần normalized vì với độ phủ 200x, ngay cả một lỗi nhỏ cũng có thể tích cộng thành điểm variant quality cao giả tạo.
- DP (Depth): số reads phủ. Quá thấp (< 8–10x) = không đủ dữ liệu thống kê; quá cao bất thường (> 3× trung bình) trong WES có thể là dấu hiệu read từ pseudogene bị align sai.
- FS (Fisher Strand bias): dùng Fisher’s exact test so sánh xem allele reference và allele alternate có phân bố đều giữa forward reads và reverse reads không. Artifact thường chỉ xuất hiện trên một chiều — khi máy đọc theo chiều đó thì lỗi xảy ra, khi đọc chiều kia thì không. Giá trị FS cao = strand bias mạnh = nghi ngờ artifact.
- ReadPosRankSum: so sánh vị trí tương đối của variant bên trong các reads mang alternate allele so với reads mang reference allele. Biến thể thật xuất hiện đồng đều từ đầu đến cuối read; artifact do lỗi sequencer thường tích trung ở đầu hoặc cuối read (nơi chất lượng thấp hơn).
- MQRankSum: so sánh mapping quality trung bình của reads mang alternate vs. reads mang reference. Nếu reads báo cáo biến thể có MAPQ thấp hơn reads báo cáo reference, gợi ý biến thể đó đến từ reads được align kém — tức là vùng lặp, và biến thể là artifact.
- SOR (Symmetric Odds Ratio): phiên bản strand bias mạnh hơn FS, đặc biệt hữu ích ở độ phủ cao nơi FS bị lạm phát thống kê.
3.3. VQSR: Học Từ Biến Thể Đã Biết
VQSR (Variant Quality Score Recalibration — tái hiệu chỉnh điểm chất lượng biến thể) là phương pháp lọc tinh vi nhất của GATK, áp dụng học máy thay vì ngưỡng cứng.
Logic cốt lõi: những biến thể trong truth set — tập biến thể đã được xác nhận độc lập bởi nhiều phương pháp (HapMap sites, 1000 Genomes high-confidence variants, dbSNP common variants) — chắc chắn là biến thể thật. Ta biết annotation profile của chúng (QD, FS, MQRankSum, ReadPosRankSum…). Ngược lại, những biến thể không có trong truth set khả năng cao là false positive. VQSR huấn luyện một Gaussian Mixture Model (GMM — một mô hình thống kê biểu diễn phân phối dữ liệu như một tập hợp các đám mây Gaussian trong không gian nhiều chiều) trên không gian annotation của true variants.
Sau khi huấn luyện, GMM tạo ra một bề mặt trong không gian annotation mô tả “vùng lãnh thổ” của true variants. Mỗi variant mới được chiếu vào không gian này và tính VQSLOD (log-odds likelihood that this variant is real). Variant nằm sâu trong vùng lãnh thổ true = VQSLOD cao = PASS; variant nằm xa rìa = VQSLOD thấp = FILTER.
Một tín hiệu chất lượng tổng thể quan trọng để chọn ngưỡng VQSLOD: Ti/Tv ratio (tỷ lệ transitions so với transversions). Trong genome người, do cơ chế deamination và các bias hóa học khác, transitions (A↔G, C↔T) xuất hiện thường xuyên hơn transversions (purine↔pyrimidine). Ti/Tv thật của WGS ~2.0–2.1; của WES coding region ~3.0. Khi filter quá nhiều true positives, Ti/Tv giảm về 1.0 (ngẫu nhiên); khi có nhiều false positives, Ti/Tv cũng lệch. Đây là một “nhiệt kế” tự nhiên để đánh giá chất lượng call set.
VQSR được chạy riêng cho SNV và indel vì chúng có error profile khác nhau. Một hạn chế: cần đủ variants (thường > 30 mẫu đa dạng) để GMM fit tốt — với cohort nhỏ hoặc single-sample phải dùng hard filtering.
3.4. Hard Filtering: Ngưỡng Cứng Phòng Khi VQSR Không Đủ Dữ Liệu
Hard filtering đặt ngưỡng cứng cho từng annotation, ví dụ: loại bỏ mọi SNV có QD < 2.0; FS > 60; MQRankSum < −12.5; ReadPosRankSum < −8.0. Variants vi phạm ≥ 1 tiêu chí sẽ có chuỗi filter tag trong cột FILTER của VCF thay vì “PASS”.
Ưu điểm: đơn giản, không cần đủ dữ liệu training. Nhược điểm: mỗi tiêu chí được áp độc lập — không học được sự tương tác giữa các annotation. Ví dụ: một variant có QD = 2.5 (vừa trên ngưỡng) nhưng cũng có FS = 55 (vừa dưới ngưỡng) sẽ PASS, nhưng kết hợp hai chỉ số “gần ngưỡng” đó là dấu hiệu đáng ngờ mà VQSR sẽ phân loại chính xác hơn.
3.5. Tín Hiệu QC Ở Cấp Độ Mẫu
Ngoài QC từng variant, cần đánh giá chất lượng tổng thể của call set từ mỗi mẫu:
- Ti/Tv ratio: như đã đề cập; kỳ vọng ~2.0–2.1 cho WGS, ~2.8–3.2 cho WES. Lệch nhiều = vấn đề với sample hoặc pipeline.
- Het/Hom ratio (tỷ lệ dị hợp tử / đồng hợp tử): ở WGS toàn genome, mỗi người mang khoảng 3–4 triệu SNV so với reference, với het/hom ≈ 1.5–2.5. Contamination làm tăng het/hom (vì biến thể của người nhiễm xuất hiện như dị hợp) hoặc làm lệch allele fraction của các het sites.
- Tỷ lệ novel variant: fraction variants không có trong dbSNP. WGS thường ~3–5% novel; WES coding region ~1–2%. Tỷ lệ novel cao bất thường = excess false positives.
- Homozygosity rate: trong nghiên cứu ung thư, loss of heterozygosity (LOH) trong tumor sample là tín hiệu sinh học quan trọng, nhưng excessive homozygosity trong germline sample gợi ý contamination hoặc inbreeding.
3.6. Preprocessing Làm Nền Tảng Cho QC
Chất lượng variant calling cuối cùng phụ thuộc không nhỏ vào hai bước tiền xử lý trước khi variant calling:
Duplicate marking: PCR khuếch đại trong library prep có thể tạo ra nhiều copies từ cùng một phân tử DNA ban đầu. Các copies này tạo ra reads giống hệt nhau (hoặc gần giống do lỗi PCR nhỏ), không phải bằng chứng độc lập. Picard MarkDuplicates hoặc samtools markdup nhận diện chúng dựa trên tọa độ 5’ của cả hai reads trong read pair — hai reads từ cùng insert sẽ có cùng tọa độ 5’. Reads trùng lặp được đánh FLAG là duplicate và loại khỏi tính toán variant calling, giữ lại chỉ một representative. Nếu không làm bước này, biến thể trong phân tử được khuếch đại nhiều nhất sẽ bị over-represented, làm lệch allele fraction và thống kê downstream.
BQSR (Base Quality Score Recalibration): máy giải trình tự tự gán base quality score cho mỗi nucleotide khi đọc. Nhưng những scores này thường không chính xác một cách có hệ thống: ví dụ, Illumina thường over-estimate quality ở giữa read và under-estimate ở cuối read; còn context như “GpCpG” có tỷ lệ lỗi cao hơn context khác. BQSR huấn luyện một mô hình hồi quy dùng các vị trí trong dbSNP là “known variants” làm reference: tại những vị trí đó, mọi sự khác biệt với reference mà không có trong dbSNP phải là lỗi kỹ thuật. Mô hình học được hàm hiệu chỉnh cho từng combination của (đọc thứ mấy, vị trí trong read, cycle, context 2-mer trước đó) → base quality chính xác hơn → variant quality scores downstream đáng tin cậy hơn.
4. Variant Annotation
4.1. Từ Tọa Độ Đến Sinh Học
Sau QC, ta có một danh sách clean variants — mỗi entry là một tuple (chromosome, position, reference allele, alternate allele, genotype). Tọa độ thuần túy này không mang ý nghĩa sinh học trực tiếp. Variant annotation là quá trình gắn thêm ngữ cảnh — nó nằm trong gene nào, ảnh hưởng đến protein thế nào, đã gặp ở bệnh nhân nào chưa, tần số trong quần thể khỏe mạnh là bao nhiêu, và các công cụ tính toán dự đoán tác động của nó là gì.
Pipeline annotation thực tế là một lớp truy vấn song song: VCF được so khớp với gene model database (Ensembl/RefSeq) để xác định vị trí trong cấu trúc gene; với nhiều cơ sở dữ liệu biến thể (dbSNP, ClinVar, gnomAD, COSMIC) để lấy thông tin hiện có; và với nhiều công cụ prediction để ước tính tác động chức năng. Kết quả là mỗi variant nhận được hàng chục trường thông tin mới.
4.2. Gene Structure Và Transcript Model
Hiểu tại sao annotation phức tạp đòi hỏi hiểu cấu trúc gene. Một gene không đơn giản là một đoạn DNA từ điểm A đến điểm B:
- Promoter: vùng upstream (trước điểm bắt đầu phiên mã), nơi các yếu tố phiên mã (transcription factors) gắn kết để điều chỉnh gene expression. Biến thể ở đây không thay đổi protein nhưng có thể thay đổi bao nhiêu protein được tạo ra.
- 5’UTR (5’ untranslated region): sau điểm bắt đầu phiên mã nhưng trước start codon. Ảnh hưởng đến hiệu quả dịch mã (translation) và độ ổn định mRNA.
- Exon: phần được dịch mã thành protein (thông qua splicing ra introns và nối exons lại).
- Intron: phần được cắt bỏ trong pre-mRNA splicing. Chứa các splice donor (GT ở đầu intron) và splice acceptor (AG ở cuối intron) — những dinucleotide này cực kỳ bảo tồn; biến thể ở đây phá vỡ splicing.
- 3’UTR: sau stop codon; chứa các vị trí gắn kết của miRNA (microRNA điều hòa gene expression) và các yếu tố ổn định/destabilize mRNA.
Phức tạp hơn nữa: hầu hết gene người có nhiều isoform (biến thể splicing) — cùng một gene có nhiều cách sắp xếp exon khác nhau cho ra các protein hơi khác nhau. Một variant tại exon chỉ có trong một số isoforms sẽ có tác động khác với variant tại exon hiện diện trong mọi isoforms. Công cụ như VEP hoặc SnpEff chú thích mỗi variant đối với mọi transcript của gene đó, sau đó xếp hạng theo “severity hierarchy” và chọn consequence nghiêm trọng nhất làm primary annotation.
4.3. Phân Loại Functional Consequence Trong Coding Region
Trong vùng mã hóa protein, hệ quả chức năng phụ thuộc vào bản chất của sự thay đổi:
Synonymous variant: thay đổi nucleotide nhưng không thay đổi amino acid. Về nguyên tắc có vẻ vô hại — nhưng không luôn trung tính. Biến thể synonymous có thể phá vỡ exonic splicing enhancer (ESE — trình tự ngắn trong exon mà protein SR-family gắn vào để tăng cường nhận diện exon trong quá trình splicing), dẫn đến exon skipping. Hoặc ảnh hưởng đến codon usage (tần số sử dụng codon ảnh hưởng tốc độ dịch mã), quan trọng trong việc gấp cuộn protein đúng thời điểm.
Missense variant: thay đổi một amino acid. Đây là loại variant phức tạp nhất để diễn giải vì tác động hoàn toàn phụ thuộc vào bối cảnh:
- Conservation (bảo tồn tiến hóa): nếu amino acid ở vị trí này giống nhau ở cá, chim, chuột, voi, và người — evolution đã “kiểm tra” biến thể ở vị trí này trong hàng trăm triệu năm và loại bỏ mọi thứ không tốt. Thay đổi nó thường nguy hiểm.
- Physicochemical similarity: thay Valine (nonpolar, nhỏ) bằng Alanine (nonpolar, nhỏ) = conservative, ít hại; thay Glycine (tiny, flexible) bằng Tryptophan (large, bulky) = drastic — có khả năng phá cấu trúc protein.
- Structural context: amino acid trong active site của enzyme hay binding interface với protein partner quan trọng hơn nhiều amino acid nằm trên một surface loop không có chức năng.
Nonsense variant: tạo ra premature stop codon. mRNA chứa stop codon sớm thường bị phân giải bởi NMD (Nonsense-Mediated mRNA Decay — cơ chế giám sát tế bào loại bỏ mRNA có stop codon sớm bất thường), dẫn đến mất protein hoàn toàn — loss of function.
Frameshift variant: insertion hoặc deletion không chia hết cho 3. Kể từ vị trí đó, tất cả codons đều bị dịch khung — protein sau đó là chuỗi amino acid vô nghĩa, thường dừng lại ở premature stop codon gần đó. Hầu như luôn loss of function.
Splice-site variant: tại canonical GT (donor) hay AG (acceptor). Splicing machinery nhận diện chính xác GT ở đầu intron và AG ở cuối; biến thể ở đây làm spliceosome bỏ qua exon (“exon skipping”) hoặc giữ lại intron trong mature mRNA. Kết quả thường tương đương hoặc tệ hơn frameshift.
Deep intronic variant: biến thể trong intron xa vị trí nối. Thường vô hại — nhưng một số tạo ra cryptic splice site (vị trí nối ẩn mà splicing machinery bắt đầu sử dụng thay thế), đưa một đoạn intron vào mRNA trưởng thành. Loại này đặc biệt khó phát hiện bằng DNA sequencing đơn thuần — cần RNA-seq để xác nhận.
4.4. Tần Số Quần Thể: Bộ Lọc Mạnh Nhất
Bộ lọc đơn giản nhưng mạnh nhất: nếu một variant xuất hiện ở tần số cao trong quần thể khỏe mạnh, nó gần như chắc chắn lành tính.
gnomAD (Genome Aggregation Database) hiện chứa >730.000 exomes và genomes từ nhiều quần thể toàn cầu, là tài nguyên tần số quần thể lớn nhất hiện tại. Logic tiến hóa đằng sau việc dùng tần số để lọc: variant gây bệnh nghiêm trọng với onset sớm không thể phổ biến trong quần thể khỏe mạnh vì natural selection loại bỏ chúng. Ngược lại, nếu 2% dân số Đông Á mang một variant mà không bị bệnh, variant đó không thể là nguyên nhân của bệnh nghiêm trọng.
Ngưỡng thực tế:
- gnomAD MAF > 1%: hầu như chắc chắn benign — BA1 criterion của ACMG
- gnomAD MAF 0.1–1%: likely benign, cần thêm bằng chứng
- gnomAD MAF < 0.01% hoặc absent: đáng được chú ý, đặc biệt với severe disease phenotype
Một lưu ý quan trọng: phải tra theo đúng population (quần thể). Một variant vắng mặt trong người châu Âu nhưng có MAF 5% trong người Đông Phi là benign, không phải pathogenic. gnomAD báo cáo tần số riêng cho từng quần thể: AFR (African), AMR (Latino), ASJ (Ashkenazi Jewish), EAS (East Asian), FIN (Finnish), NFE (Non-Finnish European), SAS (South Asian).
Một số cơ sở dữ liệu quan trọng ngoài gnomAD được tóm tắt trong bảng sau:
| Cơ sở dữ liệu | Nội dung chính | Dùng cho |
|---|---|---|
| dbSNP | rsID cho biến thể đã biết, tần số quần thể | Nhận diện biến thể phổ biến |
| ClinVar | Phân loại lâm sàng từ nhiều lab, bằng chứng pathogenicity | Clinical interpretation |
| gnomAD | Tần số allele trong >730.000 genome/exome toàn cầu | Population frequency filter |
| COSMIC | Somatic variants từ hàng triệu khối u ung thư | Cancer genomics |
| OMIM | Gene–disease associations từ y văn | Disease context |
Bảng 4.1. Các cơ sở dữ liệu cốt lõi trong variant annotation.
4.5. In Silico Predictors: Đánh Giá Biến Thể Chưa Có Tiền Lệ
Không phải mọi variant đều có sẵn trong cơ sở dữ liệu. Với các biến thể hiếm và mới, cần in silico prediction tools để ước tính tác động chức năng:
SIFT (Sorting Intolerant From Tolerant): multiple sequence alignment của protein đó với các protein tương đồng ở nhiều loài; tính tần suất mỗi amino acid xuất hiện tự nhiên ở mỗi vị trí; substitution hiếm trong evolution = likely “intolerant” = “deleterious”. Score 0.0 = deleterious; 1.0 = tolerated.
PolyPhen-2 kết hợp conservation với thông tin cấu trúc 3D: có hai mode — HumDiv (huấn luyện trên variants gây mất chức năng protein so với neutral variants trong alignment đa loài) và HumVar (huấn luyện trên clinical human disease variants) — cho ra “probably damaging”, “possibly damaging”, “benign”.
CADD (Combined Annotation Dependent Depletion): framework học máy train trên sự tương phản giữa (a) các substitution đã được quan sát và tồn tại trong quần thể người hiện tại (presumably survived selection = benign bias) và (b) các substitution ngẫu nhiên giả lập (no selection = harmful bias on average). Mô hình học cách phân biệt hai nhóm này dựa trên >60 annotation types khác nhau — conservation, gene structure, regulatory data, protein function annotations… Đầu ra là C-score Phred-scaled: CADD ≥ 20 = top 1% deleterious; ≥ 30 = top 0.1%. Lợi thế lớn của CADD: áp dụng cho mọi vùng genome (coding và non-coding) và mọi loại variant (SNV, indel).
AlphaMissense: được phát triển từ nền tảng kiến trúc của AlphaFold2 — mô hình deep learning về protein structure prediction. AlphaFold2 được train trên hàng trăm triệu protein sequences và structures đến mức model “học được” một biểu diễn phong phú về ngữ nghĩa amino acid — in effect, một protein language model. AlphaMissense fine-tunes biểu diễn này bằng dữ liệu clinical variants (pathogenic vs. benign từ ClinVar và gnomAD), tận dụng sự hiểu biết ngầm về cấu trúc protein. Kết quả: bao phủ ~216 triệu missense variants có thể có trong human genome, với độ chính xác cao hơn đáng kể so với SIFT/PolyPhen — theo benchmark, gần bằng đánh giá thực nghiệm ở nhiều gene.
Một điểm quan trọng về tất cả tools này: chúng là ước lượng xác suất, không phải chẩn đoán. Chúng có false positive và false negative rate đáng kể. Dùng chúng như một trong nhiều bằng chứng, không phải bằng chứng duy nhất.
4.6. Framework ACMG Và Phân Loại Lâm Sàng
Trong clinical genomics, annotation phải dẫn đến một phân loại lâm sàng để bác sĩ có thể đưa ra quyết định. ACMG/AMP 2015 guidelines cung cấp một framework chuẩn hóa toàn cầu với năm bậc: Pathogenic, Likely Pathogenic, VUS (Variant of Uncertain Significance), Likely Benign, Benign.
Framework ACMG không phải là điểm số đơn giản mà là một hệ thống evidence-based có cấu trúc. Mỗi loại bằng chứng được phân hạng theo sức mạnh:
Bằng chứng Pathogenic: PVS1 (predicted null variant — frameshift/nonsense/splice trong gene haploinsufficient) = rất mạnh; PS1 (cùng amino acid change đã confirmed pathogenic) = mạnh; PM1 (vị trí hotspot pathogenic trong protein) = vừa; PP3 (nhiều in silico tools đồng thuận damaging) = hỗ trợ.
Bằng chứng Benign: BA1 (MAF > 5% trong gnomAD) = standalone benign; BS1 (MAF cao hơn disease prevalence) = mạnh; BP4 (nhiều tools đồng thuận benign) = hỗ trợ.
Phân loại cuối cùng dùng logic kết hợp bằng chứng — ví dụ: 2 bằng chứng mạnh pathogenic + 1 vừa = Likely Pathogenic. Điểm mạnh của framework này là tính minh bạch và reproducibility: biết chính xác bằng chứng nào được dùng để đưa ra phân loại.
4.7. VUS Và Sự Không Chắc Chắn Là Trung Thực
Thực tế lâm sàng khó chịu: khoảng 30–50% missense variants trong clinical exome sequencing rơi vào hạng mục VUS — chúng ta không có đủ bằng chứng để phân loại dứt khoát. VUS không phải là thất bại của quy trình — nó là một phát biểu trung thực về giới hạn kiến thức hiện tại.
ClinVar là cơ chế quan trọng để reclassify VUS theo thời gian: một platform công khai nơi các phòng lab lâm sàng chia sẻ interpretations của variant. Khi 5 lab độc lập đều phân loại một variant là Pathogenic với bằng chứng consistent, một VUS có thể được reclassify. Database gnomAD tiếp tục phát triển — variant có MAF 0.005% trong gnomAD năm 2018 (và được xem là VUS) nay có thể thấy tần số 0.5% sau khi thêm 600.000 genome = likely benign. Annotation là một quá trình sống, không phải một snapshot tĩnh.
5. Bức Tranh Tổng Thể Và Xu Hướng Tương Lai
5.1. Bốn Lớp Quyết Định Lồng Nhau
Nhìn lại toàn bộ quy trình: alignment, variant calling, post-QC, và annotation không phải là bốn hộp rời rạc trong một pipeline — chúng là bốn lớp quyết định lồng nhau, mỗi lớp sử dụng output của lớp trước như nguyên liệu đầu vào và tạo ra uncertainties mới mà lớp sau phải xử lý.
Sai sót ở alignment (reads đặt nhầm vị trí do vùng lặp) → tạo ra false variant candidates → post-QC phải lọc dựa trên MAPQ thấp. Sai sót ở preprocessing (không mark duplicates tốt) → méo allele fraction → variant caller gọi sai genotype → annotation áp dụng cho genotype sai. Mỗi lỗi nhỏ ở đầu khuếch đại qua các lớp sau.
Hiểu luồng uncertainty này là kỹ năng quan trọng của người phân tích genomics: khi thấy một variant khả nghi, câu hỏi đúng không phải chỉ là “annotation cho gì?” mà là “alignment quality có ổn không? Reads phủ vị trí này có MAPQ cao không? Allele fraction có đúng kỳ vọng không? Có strand bias không?” — và sau đó mới đến annotation.
5.2. Long-Read Sequencing Thay Đổi Điều Gì
Short-read sequencing (Illumina, 150bp) là backbone của WGS hiện tại nhưng có một giới hạn cơ bản: reads ngắn hơn các vùng lặp dài → repetitive regions vĩnh viễn có MAPQ thấp → structural variants phức tạp không thể giải quyết.
Long-read sequencing từ PacBio HiFi và Oxford Nanopore Technology (ONT) đọc được 10.000–100.000 bp/read. Điều này thay đổi alignment và variant calling ở nhiều chiều:
- Reads dài hơn vùng lặp → có thể align uniquely qua vùng lặp, MAPQ cao
- Structural variants (SV — inversions, translocations, large insertions/deletions > 50bp) có thể đọc trực tiếp bằng một read thay vì phải suy luận từ split-reads và discordant pairs
- Phasing (xác định allele nào nằm trên nhiễm sắc thể nào của cặp): với reads dài, cùng một read có thể bao phủ nhiều heterozygous sites → biết chúng cùng haplotype → haplotype-resolved genome assembly có thể làm mà không cần trio sequencing
- Epigenetic modification (methyl hóa) đọc trực tiếp từ signal của ONT — không cần bisulfite treatment riêng
Thuật toán alignment và variant calling cho long reads cũng khác: minimap2 thay BWA-MEM; DeepVariant (dựa trên deep learning) tỏ ra mạnh hơn GATK cho PacBio HiFi.
5.3. Deep Learning Đang Định Hình Lại Chuẩn
DeepVariant (Google Brain, 2018) là ví dụ quan trọng nhất về deep learning applied to variant calling. Thay vì explicit statistical models như pair HMM, DeepVariant chuyển pileup tại mỗi vị trí thành một ảnh ba kênh (giống ảnh RGB) và feed vào một convolutional neural network (CNN) huấn luyện để phân loại: reference/heterozygous/homozygous alt. CNN “học” trực tiếp các pattern báo hiệu biến thể thật từ hàng triệu ví dụ training mà không cần hand-crafted rules.
Kết quả benchmarking (GIAB — Genome in a Bottle): DeepVariant có F1 score cao hơn GATK HaplotypeCaller trên Illumina data, và đặc biệt vượt trội trên long-read data. AlphaMissense (annotation), DeepVariant (calling), Parabricks (GPU-accelerated GATK) — xu hướng rõ ràng là deep learning đang thay thế hoặc nâng cấp từng module trong pipeline.
Có một điểm thú vị: deep learning models thường có better performance nhưng less interpretability — ta biết chúng đưa ra quyết định đúng hơn, nhưng khó giải thích tại sao mỗi quyết định cụ thể. Với clinical genomics nơi mỗi quyết định ảnh hưởng đến bệnh nhân, interpretability vẫn còn là một yêu cầu.
Chúng tôi sẽ đề cập quy trình thực hành — bao gồm cài đặt công cụ, cú pháp lệnh và cách giải thích kết quả — trong bài tutorial riêng.
Kết Luận
Từ 900 triệu mảnh ngắn rời rạc đến một danh sách biến thể di truyền có ý nghĩa lâm sàng là một hành trình tính toán gồm bốn lớp quyết định tinh tế, mỗi lớp xây trên nền tảng của lớp trước.
Alignment giải quyết bài toán định vị quy mô khổng lồ bằng cách xây dựng cấu trúc index BWT/FM-index từ trước, sau đó dùng chiến lược seed-and-extend để vừa nhanh vừa chính xác. Mapping quality là tín hiệu đáng tin cậy để đánh giá mức độ tự tin của từng quyết định placement.
Variant calling phân biệt tín hiệu thật khỏi nhiễu bằng cách tái lắp ráp cục bộ haplotype qua de Bruijn graph, tính likelihood qua pair HMM có tính đến base quality scores, và genotyping toàn diện bằng Bayes. Joint calling tận dụng sức mạnh của cohort để cải thiện độ chính xác cho từng mẫu.
Post-variant QC loại bỏ artifacts bằng cách nhìn đồng thời vào nhiều chiều kỹ thuật — không chỉ depth mà còn strand bias, mapping quality distribution, read position distribution — kết hợp trong VQSR hoặc hard filtering. Ti/Tv ratio và het/hom ratio là nhiệt kế tự nhiên để đánh giá chất lượng tổng thể call set.
Annotation kết nối dữ liệu cá nhân với bộ nhớ tập thể của khoa học qua cơ sở dữ liệu quần thể (gnomAD), lâm sàng (ClinVar), và ung thư (COSMIC); dự đoán tác động bằng các lớp công cụ từ SIFT đến AlphaMissense; và cuối cùng dẫn đến phân loại ACMG có cấu trúc evidence-based.
Xu hướng tương lai — long reads tăng độ phân giải vùng lặp và structural variants; deep learning thay thế dần các mô hình thống kê tường minh — đang nhanh chóng mở rộng khả năng của từng bước. Nhưng bất kể công cụ nào được dùng, trực giác về tại sao mỗi bước tồn tại và điểm nào mỗi bước có thể thất bại là điều kiện tiên quyết để phân tích đáng tin cậy và diễn giải kết quả có trách nhiệm.
Tài Liệu Tham Khảo
Li, H., & Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Aligner. Bioinformatics, 25(14), 1754–1760. https://doi.org/10.1093/bioinformatics/btp324
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–1303. https://doi.org/10.1101/gr.107524.110
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., Kling, D. E., Gauthier, L. D., Levy-Moonshine, A., Roazen, D., Shakir, K., Thibault, J., Chandramouliswaran, I., Gabriel, S., Altshuler, D., Daly, M., & Bank, E. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. https://doi.org/10.1101/201178
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R. S., Thormann, A., Flicek, P., & Cunningham, F. (2016). The Ensembl Variant Effect Predictor. Genome Biology, 17(1), 122. https://doi.org/10.1186/s13059-016-0974-4
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. https://doi.org/10.1038/gim.2015.30
Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., Pritzel, A., Wong, L. H., Zuk, O., Jumper, J., Hassabis, D., Kohli, P., & Žemgulytė, A. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science, 381(6664), eadg7492. https://doi.org/10.1126/science.adg7492
Poplin, R., Chang, P.-C., Alexander, D., Schwartz, S., Colthurst, T., Ku, A., Newburger, D., Dijamco, J., Nguyen, N., Afshar, P. T., Gross, S. S., Dahl, L., DePristo, M. A., & DePristo, M. A. (2018). A universal SNP and small-indel variant caller using deep neural networks. Nature Biotechnology, 36(10), 983–987. https://doi.org/10.1038/nbt.4235
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., Collins, R. L., Laricchia, K. M., Ganna, A., Birnbaum, D. P., Gauthier, L. D., Brand, H., Solomonson, M., Watts, N. A., Rhodes, D., Singer-Berk, M., England, E. M., Seaby, E. G., Kosmicki, J. A., … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. https://doi.org/10.1038/s41586-020-2308-7
Explore more like this
Gene Regulatory Network: Từ Phân Tử Đến Multi-Omics Và Ứng Dụng Lâm Sàng
Genome của người chứa khoảng 20.000 gene mã hóa protein, nhưng không phải gene nào cũng hoạt động ở mọi tế bào trong mọi thời điểm. Một tế bào gan,...
Transcriptomics: Hành Trình Giải Mã Hệ Phiên Mã và Các Công Nghệ Chủ Chốt
Tế bào người chứa khoảng 3,2 tỷ cặp bazơ DNA — nhưng không phải tất cả đều được “đọc” cùng một lúc, hay ở mọi loại tế bào. Một tế...
Single-Cell Omics: Khi Khoa Học Nhìn Vào Từng Tế Bào
Hãy tưởng tượng bạn muốn hiểu một thành phố bằng cách hỏi một câu hỏi duy nhất với tất cả 10 triệu cư dân cùng lúc và lấy trung bình...
Comments