Các Công Nghệ Đọc Genome: Từ Sanger Đến Long-Read và Sự Tiến Hoá Qua Các Thế Hệ


Năm 1977, Frederick Sanger công bố một phương pháp cho phép con người lần đầu tiên “đọc” trình tự DNA theo từng ký tự nucleotide. Thủ công, chậm chạp, và đòi hỏi kỹ năng thực nghiệm cao, nhưng nguyên lý của nó đặt nền móng cho genomics hiện đại. Gần năm thập kỷ sau, các máy giải trình tự xách tay nhỏ hơn điện thoại thông minh có thể đọc hàng gigabase DNA ngay tại rừng Amazon hoặc phòng điều trị bệnh viện. Khoảng cách giữa hai mô tả đó không phải là sự cải tiến đơn tuyến mà là cả một chuỗi các cuộc cách mạng công nghệ xảy ra theo nối tiếp, mỗi lần làm thay đổi hoàn toàn câu hỏi mà nhà khoa học có thể đặt ra. Bài viết này truy xuất toàn bộ hành trình đó: từ phương pháp Sanger qua vi mảng SNP, đến giải trình tự thế hệ kế tiếp (NGS) đọc ngắn, và cuối cùng là công nghệ đọc dài hiện tại.
1. Câu Hỏi Trung Tâm
1.1. Ba Câu Hỏi Sinh Học Nền Tảng
Để hiểu tại sao nhiều công nghệ cùng tồn tại, cần bắt đầu từ các câu hỏi sinh học. Genomics không phải là một câu hỏi duy nhất; nó là một tập hợp các câu hỏi ở các cấp độ phân giải khác nhau:
Câu hỏi 1 — Trình tự là gì? Trình tự DNA của sinh vật này, vùng gene này, hay cá thể này trông như thế nào? Đây là câu hỏi đòi hỏi phương pháp giải trình tự.
Câu hỏi 2 — Biến thể là gì? Trình tự của cá thể này khác với tham chiếu hoặc cá thể khác ở những vị trí nào? Đây là câu hỏi di truyền quần thể và y học chính xác.
Câu hỏi 3 — Cấu trúc và ngữ cảnh là gì? DNA gấp, sắp xếp, và hoạt động như thế nào ở cấp độ toàn bộ nhiễm sắc thể? Đây là câu hỏi đòi hỏi phân giải cấu trúc.
Mỗi thế hệ công nghệ genomics đã mở rộng khả năng trả lời từng loại câu hỏi này theo những cách khác nhau. Bảng 1.1 tóm tắt ánh xạ tổng quát.
| Câu hỏi | Công nghệ chính | Hạn chế của thế hệ trước |
|---|---|---|
| Trình tự cụ thể, chính xác cao | Sanger sequencing | Thông lượng thấp, không thể mở rộng |
| Biến thể phổ biến trong quần thể | Vi mảng SNP / GWAS | Chỉ phát hiện được biến thể đã biết trước |
| Toàn bộ genome, độ sâu cao | Short-read NGS (Illumina) | Không đọc được vùng lặp dài |
| Cấu trúc, haplotype, methylation | Long-read (PacBio, Nanopore) | Chi phí trên mỗi base còn cao hơn NGS |
Bảng 1.1. Ba câu hỏi sinh học nền tảng và công nghệ tiêu biểu giải quyết từng loại.
1.2. Lý Do Không Có “Công Nghệ Tất Cả Trong Một”
Mỗi công nghệ đánh đổi giữa ba trục: độ chính xác (accuracy), độ dài đọc (read length), và thông lượng (throughput). Không có công nghệ nào tối ưu cả ba cùng lúc. Sanger cho độ chính xác và độ dài đọc tốt nhưng thông lượng thấp; Short-read NGS cho thông lượng và độ chính xác cao nhưng đọc ngắn; Long-read cho độ dài đọc lớn nhưng lịch sử đánh đổi với độ chính xác mỗi lần đọc. Hiểu tam giác đánh đổi này là chìa khóa để lựa chọn công nghệ phù hợp cho từng câu hỏi nghiên cứu (xem Hình 1.1).
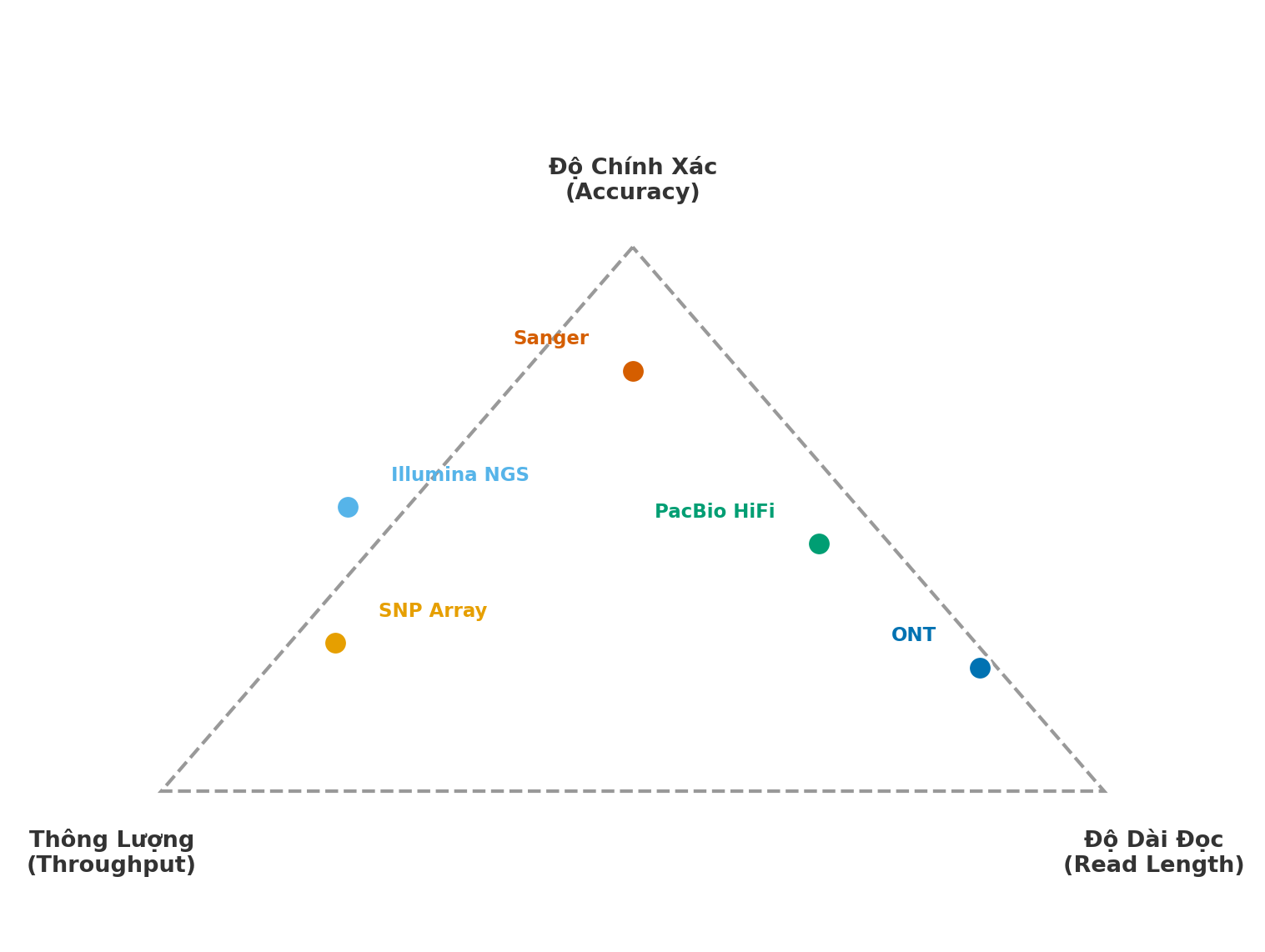 Hình 1.1. Tam giác đánh đổi giữa độ chính xác, độ dài đọc và thông lượng cho ba thế hệ công nghệ chính.
Hình 1.1. Tam giác đánh đổi giữa độ chính xác, độ dài đọc và thông lượng cho ba thế hệ công nghệ chính.
2. Kỷ Nguyên Sanger
2.1. Nguyên Lý Phương Pháp Sanger
Giải trình tự Sanger (Sanger sequencing, còn gọi là phương pháp dideoxy hay chain-termination sequencing) dựa trên một cơ chế tinh tế: sử dụng các phân tử dideoxynucleotide (ddNTP, nucleotide thiếu nhóm hydroxyl -OH ở vị trí 3’ của đường ribose) làm “điểm dừng” ngẫu nhiên trong quá trình tổng hợp DNA.
Khi DNA polymerase kéo dài một chuỗi DNA mới và vô tình gắn một ddNTP thay vì một dNTP bình thường, quá trình kéo dài bị chấm dứt vì không có nhóm -OH 3’ để hình thành liên kết phosphodiester tiếp theo. Bằng cách thực hiện bốn phản ứng song song, mỗi phản ứng chứa một loại ddNTP (ddATP, ddCTP, ddGTP hoặc ddTTP) được đánh dấu phóng xạ (hoặc huỳnh quang trong phiên bản hiện đại), kết quả thu được là một tập hợp các đoạn DNA có độ dài khác nhau, mỗi đoạn kết thúc tại một loại nucleotide cụ thể. Điện di gel phân tách các đoạn theo kích thước, và kết quả đọc từ dưới lên trên của gel cho ra trình tự nucleotide (Sanger et al., 1977).
Phương pháp này ban đầu đòi hỏi gel polyacrylamide và film X-quang để phát hiện phóng xạ. Bước ngoặt đầu tiên đến vào năm 1986 khi Applied Biosystems ra mắt máy giải trình tự tự động ABI 370, thay thế phóng xạ bằng bốn màu huỳnh quang khác nhau cho bốn loại ddNTP, cho phép chạy một phản ứng duy nhất thay vì bốn phản ứng riêng biệt. Tự động hoá biến việc đọc trình tự từ kỹ năng thủ công của chuyên gia thành quy trình có thể nhân rộng.
2.2. Dự Án Human Genome và Giới Hạn Của Sanger
Dự án Human Genome (Human Genome Project, HGP) khởi động năm 1990 như một nỗ lực quốc tế nhằm xác định trình tự toàn bộ khoảng 3,2 tỷ cặp base của human genome. HGP hoàn toàn dựa vào giải trình tự Sanger và trở thành công trình khoa học hợp tác lớn nhất trong lịch sử sinh học, kéo dài 13 năm và tiêu tốn khoảng 3 tỷ USD trước khi công bố bản thảo vào năm 2000 và bản hoàn chỉnh vào năm 2003 (International Human Genome Sequencing Consortium, 2004).
Thành tựu này bộc lộ rõ ràng cả sức mạnh lẫn giới hạn của Sanger. Sức mạnh: độ chính xác trên 99,999% và khả năng đọc các đoạn 500-1000 nucleotide với chất lượng cao. Giới hạn: để giải trình tự một human genome, cần thực hiện hàng triệu phản ứng riêng lẻ, đòi hỏi ít nhất hàng chục ngàn máy chạy song song trong nhiều năm. Chi phí giải trình tự một human genome vào năm 2001 ước tính khoảng 95 triệu USD. Rõ ràng, genomics quy mô lớn cần một mô hình công nghệ hoàn toàn khác.
Một điểm quan trọng cần nhấn mạnh là ngay cả genome “hoàn chỉnh” từ HGP năm 2003 vẫn còn những khoảng trống (gap) tại một số vùng dị nhiễm sắc (heterochromatin: vùng DNA cuộn chặt, giàu trình tự lặp, khó giải trình tự). Các khoảng trống này không được lấp đầy hoàn toàn cho đến năm 2022 khi dự án Telomere-to-Telomere (T2T) công bố human genome đầu tiên thực sự hoàn chỉnh từ đầu đến cuối mỗi nhiễm sắc thể, nhờ vào công nghệ long-read (Nurk et al., 2022).
3. Vi Mảng DNA và Kỷ Nguyên SNP Array
3.1. Nguyên Lý Hybridization
Trong khi giải trình tự Sanger đọc DNA từng nucleotide một, vi mảng DNA (DNA microarray) tiếp cận bài toán theo hướng hoàn toàn khác: thay vì đọc mới, chúng hỏi xem DNA mẫu có khớp với một tập hợp trình tự đã biết từ trước hay không.
Nguyên lý cốt lõi là hybridization (lai phân tử): hai sợi DNA đơn có trình tự bổ sung sẽ tự động kết cặp theo nguyên tắc Watson-Crick. Một vi mảng DNA là một bề mặt nhỏ (thường là kính hoặc silicon) trên đó hàng nghìn đến hàng triệu đoạn DNA ngắn gọi là probe (đầu dò) được gắn cố định tại các vị trí xác định. DNA mẫu của cá thể cần khảo sát được cắt thành các đoạn nhỏ, đánh dấu huỳnh quang, rồi cho tiếp xúc với bề mặt chip. Nếu đoạn DNA mẫu có trình tự bổ sung với một probe, nó sẽ lai vào probe đó. Sau khi rửa loại bỏ DNA không lai, mức độ huỳnh quang tại mỗi vị trí phản ánh mức độ hybridization và từ đó cho biết cá thể có mang trình tự tương ứng hay không.
Đây là hướng tiếp cận kiến thức có sẵn trước (prior knowledge approach): vi mảng không thể phát hiện điều gì mới mẻ hoàn toàn; nó chỉ có thể xác nhận hoặc phủ nhận sự hiện diện của những trình tự đã được đưa vào chip trong quá trình thiết kế.
3.2. Từ Expression Array Đến SNP Array
Expression microarray (DNA expression microarray) là ứng dụng đầu tiên, ra đời từ bài báo công bố năm 1995 của Schena và cộng sự, trong đó họ sử dụng vi mảng để đo đồng thời gene expression của khoảng 45 gene từ Arabidopsis thaliana (Schena et al., 1995). Ý tưởng này nhanh chóng mở rộng, và đến cuối thập kỷ 1990, expression microarray với hàng chục nghìn probe trở thành công cụ tiêu chuẩn để so sánh gene expression profile giữa các điều kiện thí nghiệm.
SNP array (vi mảng đa hình nucleotide đơn) là đặc biệt hóa của vi mảng để phát hiện các SNP (Single Nucleotide Polymorphism, đa hình nucleotide đơn: sự khác biệt ở một nucleotide duy nhất tại một vị trí trên genome giữa các cá thể trong quần thể). Affymetrix ra mắt các chip SNP đầu tiên vào cuối thập kỷ 1990, và Illumina phát triển dòng BeadChip của họ vào đầu thập kỷ 2000. Những chip hiện đại nhất có thể đồng thời genotype hơn 5 triệu vị trí SNP từ một mẫu DNA duy nhất với chi phí dưới 100 USD.
Tại sao focus vào SNP? Human genome có khoảng 4-5 triệu vị trí SNP khác biệt so với reference genome GRCh38. Phần lớn biến thể di truyền giữa các cá thể người nằm ở những vị trí này. Hơn nữa, do liên kết không cân bằng (linkage disequilibrium, LD: xu hướng của các allele gần nhau trên cùng một nhiễm sắc thể di truyền cùng nhau qua các thế hệ), việc genotype một tập hợp chiến lược các SNP “đại điện” có thể cho phép suy luận trạng thái của nhiều SNP khác ở xung quanh mà không cần đo trực tiếp. Đây là nguyên tắc thống kê gọi là imputation (suy giải: quá trình ước lượng trạng thái các biến thể di truyền chưa được đo dựa trên mối tương quan cấu trúc với các biến thể đã được đo).
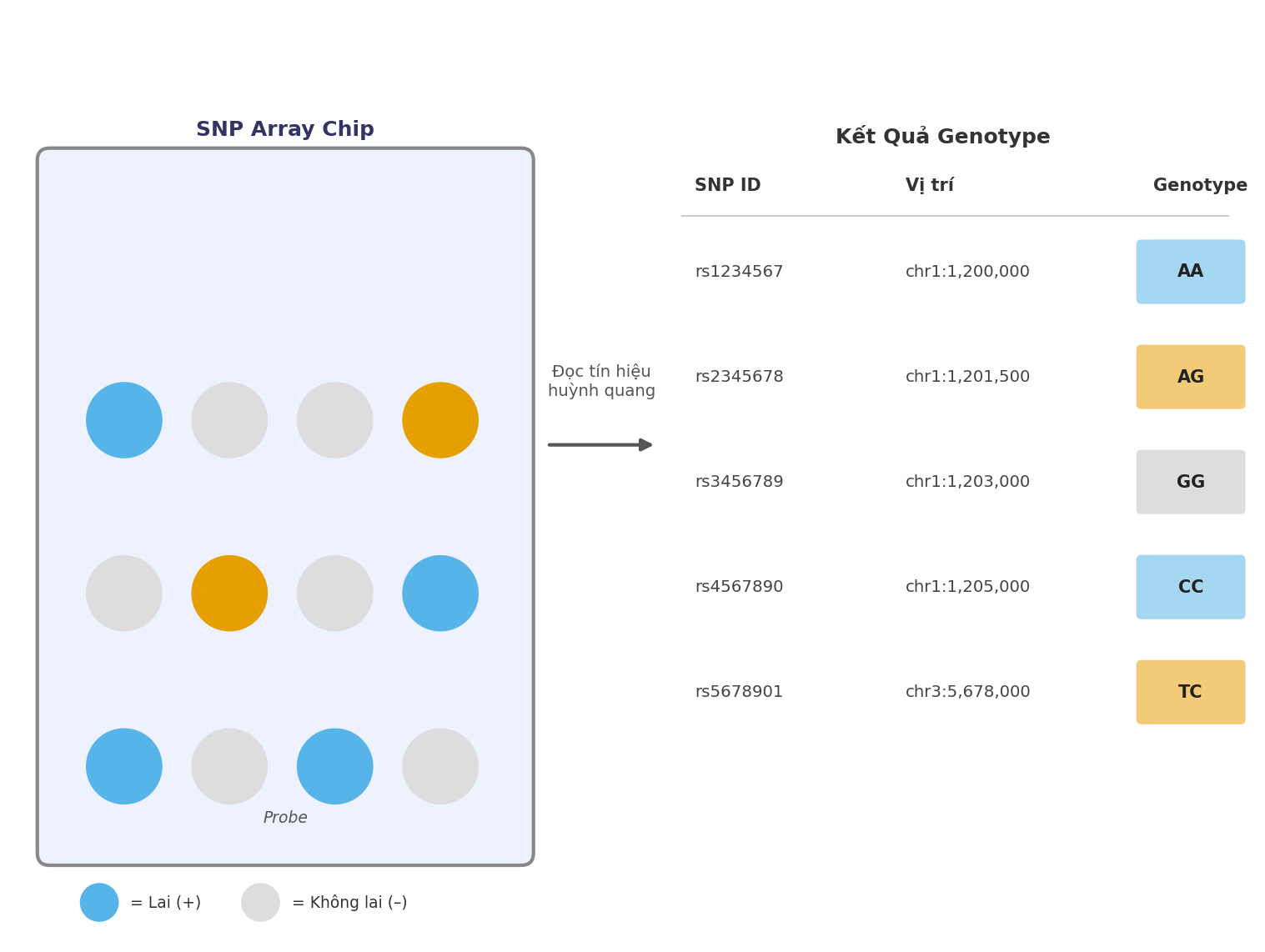 Hình 3.1. Nguyên lý hoạt động của SNP array: probe gắn trên chip lai với DNA mẫu; tín hiệu huỳnh quang tại mỗi vị trí cho biết allele tại SNP đó.
Hình 3.1. Nguyên lý hoạt động của SNP array: probe gắn trên chip lai với DNA mẫu; tín hiệu huỳnh quang tại mỗi vị trí cho biết allele tại SNP đó.
3.3. GWAS và Câu Hỏi Di Truyền Học Quần Thể
GWAS (Genome-Wide Association Study, nghiên cứu kết hợp toàn bộ genome) là ứng dụng khoa học quan trọng nhất của SNP array. Trong một GWAS, hàng nghìn đến hàng chục nghìn cá thể được chia thành nhóm bệnh và nhóm đối chứng; tất cả đều được genotype bằng SNP array; sau đó phân tích thống kê tìm kiếm các vị trí SNP có tần số allele khác biệt đáng kể giữa hai nhóm. Những vị trí như vậy gọi là loci liên kết (associated loci), cho thấy chúng có liên quan đến nguy cơ mắc bệnh.
Năm 2007, Wellcome Trust Case Control Consortium (WTCCC) thực hiện một trong những GWAS quy mô nhất thời đó, phân tích 14.000 trường hợp bệnh cho bảy bệnh thường gặp (tiểu đường type 1, tiểu đường type 2, bệnh tim mạch vành, bệnh Crohn, tăng huyết áp, viêm khớp dạng thấp, và bệnh lưỡng cực) cùng 3.000 đối chứng dùng chung. Nghiên cứu xác định hàng chục loci có ý nghĩa thống kê mới, thay đổi cách hiểu về cơ sở di truyền của các bệnh phức tạp (Wellcome Trust Case Control Consortium, 2007).
Tuy nhiên, GWAS và SNP array có những hạn chế cơ bản. Thứ nhất, SNP array chỉ phát hiện được các biến thể đã biết trước và có tần số đủ cao trong quần thể. Các biến thể hiếm (rare variant: biến thể có tần số allele thứ hai dưới 1-5% trong quần thể) thường không được đưa vào chip hoặc không đủ lực thống kê để phát hiện. Thứ hai, SNP array không phát hiện được các biến thể cấu trúc (structural variant: các thay đổi DNA lớn như mất đoạn, nhân đoạn, đảo đoạn hoặc di chuyển vị trí). Thứ ba, kết quả GWAS chỉ cho biết vị trí thống kê chứ không chỉ ra trực tiếp gene hay cơ chế nhân quả.
4. Thế Hệ Thứ Hai: Short-Read NGS
4.1. Triết Lý Sequencing By Synthesis
Sự kiện thay đổi cuộc chơi xảy ra vào năm 2004-2005 khi 454 Life Sciences (sau đó thuộc Roche) ra mắt nền tảng giải trình tự thế hệ tiếp theo đầu tiên thương mại, dựa trên pyrosequencing (Margulies et al., 2005). Thay vì giải trình tự từng đoạn DNA riêng lẻ như Sanger, 454 thực hiện hàng triệu phản ứng song song trong những giếng picolitre cực nhỏ. Lần đầu tiên thuật ngữ massively parallel sequencing (giải trình tự song song đại trà) trở nên có ý nghĩa thực tiễn.
Illumina thực sự thống trị thị trường NGS sau khi mua lại công ty Solexa năm 2006 và ra mắt Illumina Genome Analyzer năm 2007. Công nghệ cốt lõi của Illumina là Sequencing by Synthesis (SBS, giải trình tự bằng tổng hợp): DNA mẫu được cắt ngẫu nhiên, gắn adapter ở hai đầu, rồi khuếch đại tạo thành các cluster (cụm: hàng nghìn bản copy của cùng một đoạn DNA gắn trên bề mặt flow cell). DNA polymerase kéo dài sợi mới sử dụng các reversible terminator nucleotide (nucleotide dừng có thể tháo được: dNTP được gắn thêm nhóm hoá học chặn sự kéo dài và nhóm huỳnh quang đặc trưng). Sau mỗi chu kỳ gắn một nucleotide, camera chụp ảnh toàn bộ flow cell để ghi nhận màu huỳnh quang tại mỗi cluster, sau đó nhóm chặn bị loại bỏ để cho phép chu kỳ tiếp theo. Quá trình này lặp đi lặp lại vài trăm lần để đọc những đoạn DNA 100-300 bp (Bentley et al., 2008).
Độ chính xác lý thuyết của phương pháp này rất cao (> 99,9% trên một nucleotide) vì tín hiệu đến từ cả một cluster gồm hàng nghìn phân tử, không phải từ một phân tử đơn. Đây là điểm khác biệt cơ bản so với công nghệ third-generation sẽ thảo luận sau.
4.2. Ứng Dụng Và Thông Lượng
Sức mạnh thực sự của Illumina nằm ở khả năng đẩy thông lượng lên vượt bậc. Mô hình HiSeq 2000 ra mắt năm 2010 có thể giải trình tự một human genome với độ sâu 30x (mỗi vị trí được đọc trung bình 30 lần) trong vài ngày với chi phí dưới 10.000 USD, giảm từ hàng triệu USD của thời Sanger. Năm 2023, NovaSeq X Plus tiến đến ngưỡng dưới 200 USD cho một human genome toàn phần.
Nhờ thông lượng cao, short-read NGS không chỉ giải quyết câu hỏi về biến thể di truyền mà còn mở ra một loạt ứng dụng mới:
WGS (Whole Genome Sequencing, giải trình tự toàn bộ genome): thay thế SNP array khi cần phát hiện cả biến thể phổ biến lẫn biến thể hiếm.
WES (Whole Exome Sequencing, giải trình tự toàn bộ exome: 1-2% của genome mã hoá protein): tiết kiệm chi phí hơn WGS khi câu hỏi tập trung vào biến thể protein-coding.
RNA-seq (giải trình tự transcriptome): cDNA được tổng hợp từ RNA rồi giải trình tự bằng NGS, cho phép đo mức gene expression với độ phân giải cao hơn nhiều so với expression microarray.
ChIP-seq (Chromatin Immunoprecipitation Sequencing): xác định vị trí gắn kết của protein điều hoà hoặc dấu hiệu histone trên toàn bộ genome.
ATAC-seq (Assay for Transposase-Accessible Chromatin Sequencing): lập bản đồ các vùng chromatin “mở” là nơi các nhân tố phiên mã có thể tiếp cận.
4.3. Câu Hỏi Mà NGS Giải Đáp Và Hạn Chế Còn Lại
Short-read NGS đã giải quyết câu hỏi thứ nhất và thứ hai một cách triệt để: ai có thể giải trình tự toàn bộ human genome với chi phí tiếp cận, phát hiện hầu hết các biến thể nucleotide. Tuy nhiên, câu hỏi thứ ba vẫn nằm ngoài tầm với.
Vấn đề căn bản của short-read là đoạn đọc quá ngắn để giải quyết các vùng DNA lặp (repeat: các đoạn trình tự giống nhau hoặc rất tương đồng xuất hiện nhiều lần trong genome). Human genome chứa nhiều loại trình tự lặp: Alu element (~300 bp), LINE-1 (~6 kb), vùng telomere, và các trình tự lặp tandem. Khi một đoạn 150 bp đọc trùng với một vùng mà cùng một trình tự xuất hiện 100 lần ở 100 vị trí khác nhau trên genome, thuật toán lắp ghép (assembly) không thể xác định đoạn đọc đó thuộc vị trí nào. Tương tự, structural variant (SV) bao gồm các xóa đoạn, nhân đoạn, và đảo đoạn lớn thường liên quan trực tiếp đến bệnh lý nhưng cần đọc dài hàng chục kilobase để phát hiện và xác định ranh giới chính xác (xem Hình 4.1).
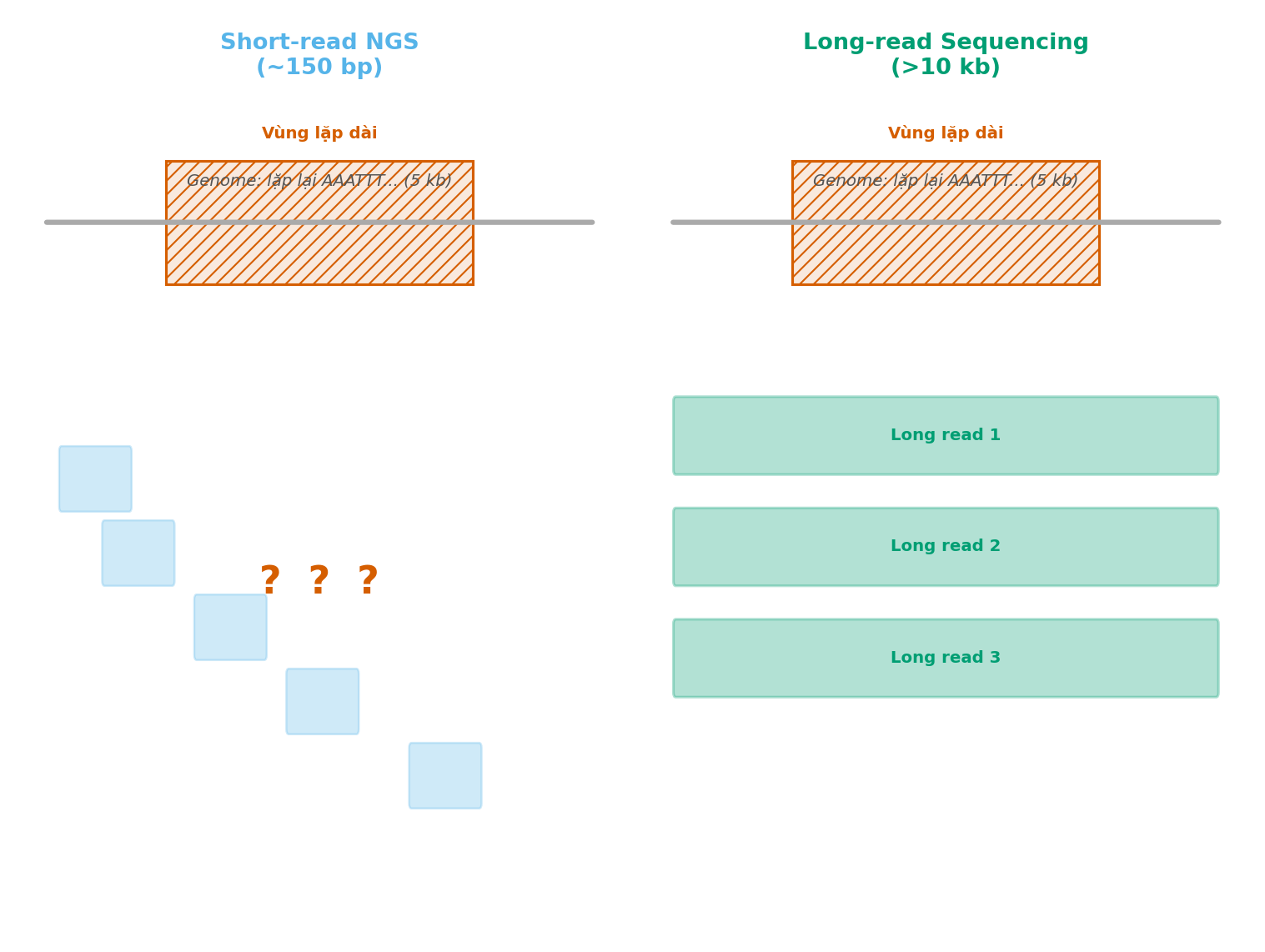 Hình 4.1. Giới hạn của short-read khi đối mặt với vùng DNA lặp dài so với long-read có thể kéo dài qua toàn bộ vùng lặp.
Hình 4.1. Giới hạn của short-read khi đối mặt với vùng DNA lặp dài so với long-read có thể kéo dài qua toàn bộ vùng lặp.
5. Thế Hệ Thứ Ba và Tư: Long-Read Sequencing
5.1. Pacific Biosciences và SMRT Sequencing
Pacific Biosciences (PacBio) thương mại hoá nền tảng SMRT (Single Molecule Real Time, giải trình tự thời gian thực phân tử đơn) vào năm 2011. Khác với Illumina dựa vào khuếch đại cụm, SMRT quan sát trực tiếp một phân tử DNA polymerase duy nhất đang tổng hợp DNA trong thời gian thực.
Cấu trúc cốt lõi là ZMW (Zero-Mode Waveguide, ống dẫn sóng không-mode-số-không: một giếng kim loại có đường kính ~70 nm, nhỏ đến mức ánh sáng không thể truyền qua theo mode thông thường, tạo ra vùng chiếu sáng cực nhỏ chỉ ở đáy giếng). Một phân tử DNA polymerase được cố định ở đáy ZMW. Khi polymerase gắn mỗi dNTP huỳnh quang vào sợi mới, nucleotide nằm trong vùng chiếu sáng và phát huỳnh quang đặc trưng trong một phần nghìn giây trước khi nhóm huỳnh quang bị cắt bỏ và sợi tiếp tục kéo dài. Camera tốc độ cao ghi nhận từng sự kiện gắn nucleotide theo thời gian, từ đó suy ra trình tự (Eid et al., 2009).
Phiên bản đầu tiên (CLR, Continuous Long Read) có độ dài đọc trung bình hàng chục kilobase nhưng tỷ lệ lỗi ~10-15% do đặc tính ngẫu nhiên của đọc phân tử đơn. Bước nhảy vọt đến với chế độ HiFi (CCS, Circular Consensus Sequencing): DNA mẫu được ligase thành vòng tròn, và polymerase đọc vòng đó nhiều lần. Trung bình tất cả các lần đọc của cùng một phân tử vòng tròn cho ra một HiFi read với độ chính xác >99,9% (Q30 trở lên) trong khi vẫn giữ được độ dài đọc 10-20 kb. HiFi đã thay đổi cục diện cạnh tranh: lần đầu tiên long-read đạt được độ chính xác tương đương short-read Illumina.
5.2. Oxford Nanopore và Đọc Điện Hoá Trực Tiếp
Oxford Nanopore Technologies (ONT) theo một nguyên lý hoàn toàn khác. Thay vì quan sát huỳnh quang từ polymerase, ONT luồn sợi DNA đơn qua một protein nanopore (một kênh protein dạng nanopore cắm trong màng polyme) và đo sự thay đổi dòng điện ion khi các nucleotide khác nhau đi qua lòng kênh.
Mỗi nucleotide (hoặc tổ hợp k-mer gồm 5-6 nucleotide nằm trong lòng pore tại một thời điểm) gây ra một mức cản trở dòng điện đặc trưng. Thuật toán basecalling (gọi base: chuyển đổi tín hiệu dòng điện thô thành trình tự nucleotide) sử dụng mạng nơ-ron học sâu để giải mã tín hiệu này thành trình tự. Tốc độ đọc vào khoảng 400-1000 bp/giây mỗi nanopore, và một chip MinION có vài nghìn kênh hoạt động đồng thời.
Những điểm độc đáo của ONT so với các nền tảng khác:
Độ dài đọc không giới hạn về lý thuyết: đã có báo cáo các đọc dài hơn 2 Mb (2 triệu bp) trong các thí nghiệm ultra-long read, dù đây là ngoại lệ. Độ dài đọc trung bình N50 trong thí nghiệm thường là 20-50 kb, đủ để bao phủ hầu hết các cấu trúc lặp trong human genome.
Phát hiện base modification trực tiếp: tín hiệu dòng điện thay đổi khi đi qua 5-methylcytosine (5mC, cytosine bị methyl hoá) khác với cytosine thông thường. Điều này cho phép ONT đọc đồng thời trình tự DNA và trạng thái methylation (methyl hoá: một dạng biến đổi epigenetic trong đó nhóm methyl được thêm vào cytosine, ảnh hưởng đến gene expression) mà không cần quy trình bisulfite-sequencing tốn kém.
Thiết bị siêu di động: Máy MinION nặng chỉ 90 gram, kết nối qua USB, và có thể dùng pin laptop để giải trình tự ngay tại thực địa. Trong đại dịch COVID-19, MinION được triển khai để giải trình tự SARS-CoV-2 ở những vùng không có phòng thí nghiệm hiện đại (Loman et al., 2015 đã chứng minh tiền đề này với dịch Ebola).
5.3. T2T và Hệ Gen Người Hoàn Chỉnh Thực Sự
Sự kết hợp giữa PacBio HiFi và ONT ultra-long read là điều kiện cần thiết để hoàn thành dự án Telomere-to-Telomere (T2T). Năm 2022, nhóm nghiên cứu do Adam Phillippy lãnh đạo công bố human genome đầu tiên không có khoảng trống, bao gồm cả hai vùng telomere (chuỗi lặp tandem TTAGGG bảo vệ đầu nhiễm sắc thể) và centromere (vùng thắt trung tâm nhiễm sắc thể chứa hàng triệu base lặp alpha satellite), những vùng mà short-read Illumina không thể giải quyết trong gần 20 năm (Nurk et al., 2022). Genome T2T-CHM13 bổ sung thêm khoảng 200 Mb trình tự mới so với GRCh38, phần lớn là vùng dị nhiễm sắc trước đây chưa được đặc trưng.
6. Đường Thời Gian và Sự Tiến Hoá
6.1. Bốn Kỷ Nguyên Công Nghệ
Nhìn lại hành trình từ 1977, có thể phân chia lịch sử genomics thành bốn kỷ nguyên rõ rệt, mỗi kỷ nguyên được định nghĩa bởi một ngưỡng thông lượng và một bộ câu hỏi mới trở nên khả thi (xem Hình 6.1).
Kỷ nguyên Sanger (1977-2004): đọc cụ thể, chính xác cao, thông lượng thấp. Câu hỏi khả thi: trình tự của một gene cụ thể là gì? Ứng dụng cline: chẩn đoán bệnh đơn gene, nghiên cứu cơ bản.
Kỷ nguyên Microarray (1995-2010): song song nhưng chỉ biến thể đã biết. Câu hỏi khả thi: trong số hàng triệu biến thể đã biết, cá thể này mang những biến thể nào? Ứng dụng chính: GWAS, phân loại ung thư dựa trên hồ sơ gene expression.
Kỷ nguyên Short-read NGS (2005-nay): thông lượng cao, đọc ngắn. Câu hỏi khả thi: toàn bộ biến thể nucleotide trong genome; gene expression toàn genome; vị trí gắn protein điều hoà. Ứng dụng chính: WGS, RNA-seq, epigenomics.
Kỷ nguyên Long-read (2011-nay): đọc dài, cấu trúc chromatin, methylation trực tiếp. Câu hỏi khả thi: cấu trúc genome hoàn chỉnh, haplotype, structural variant, pangenome.
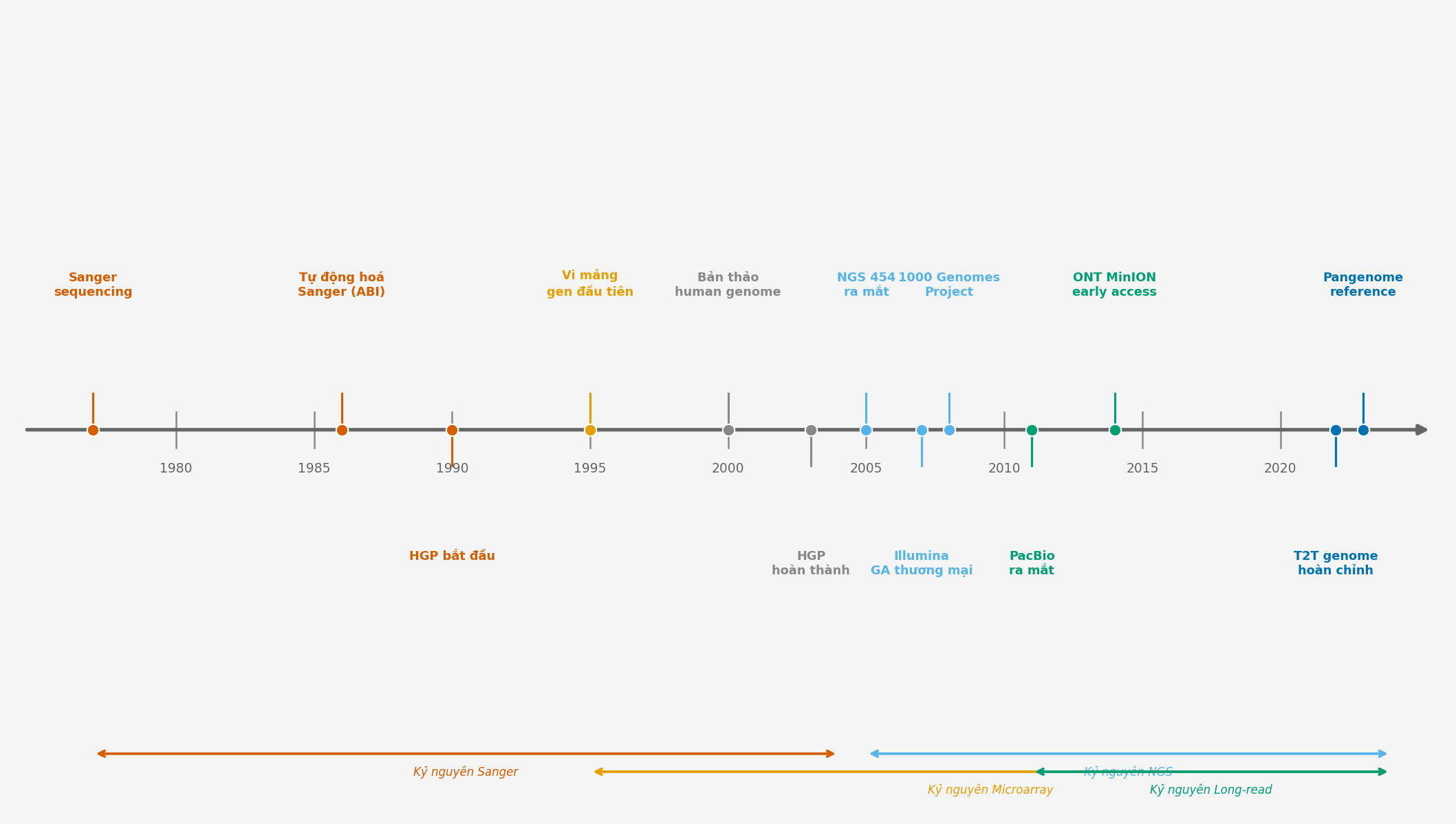 Hình 6.1. Đường thời gian các cột mốc quan trọng trong lịch sử công nghệ genomics từ 1977 đến 2023.
Hình 6.1. Đường thời gian các cột mốc quan trọng trong lịch sử công nghệ genomics từ 1977 đến 2023.
6.2. Quy Luật Moore Của Genomics
Một trong những đặc điểm ấn tượng nhất của lịch sử genomics là tốc độ giảm chi phí vượt xa cả định luật Moore trong công nghiệp bán dẫn. Chi phí giải trình tự một human genome đã giảm từ ~95 triệu USD năm 2001 xuống ~1.000 USD vào năm 2015 và xuống dưới 200 USD năm 2023, tức là giảm khoảng 500.000 lần trong 22 năm. Trong khoảng thời gian tương đương, sức mạnh tính toán theo định luật Moore chỉ tăng khoảng 1.000 lần. Điều này được thực hiện thông qua sự kết hợp của cải tiến hoá học (phản ứng phân tích hiệu quả hơn), cải tiến quang học (camera độ phân giải cao hơn), cải tiến sinh hoá (polymerase chính xác hơn), và đặc biệt là thiết kế chip flow cell mật độ cao hơn.
Biểu đồ “Wetterstrand curve” (đường cong giảm chi phí giải trình tự do NHGRI theo dõi) là tài liệu tham chiếu tiêu chuẩn cho thấy sự giảm này. Bước ngoặt rõ ràng nhất xảy ra vào khoảng 2007-2008 khi NGS thế hệ hai đột ngột làm chi phí giảm dốc xa hơn dự đoán của định luật Moore.
7. So Sánh Tổng Hợp
7.1. Ma Trận Đặc Tính Kỹ Thuật
Bảng 7.1 so sánh chi tiết các đặc tính kỹ thuật và ứng dụng của các nền tảng chính, dựa trên thông số hiện tại (2024-2025).
| Đặc tính | Sanger | SNP Array | Illumina NGS | PacBio HiFi | ONT |
|---|---|---|---|---|---|
| Độ dài đọc | 500-1000 bp | N/A | 100-300 bp | 15-25 kb | 10 kb-2 Mb |
| Độ chính xác/lần đọc | >99,99% | >99,9% | ~99,9% | >99,5% | ~95-99% (Q20) |
| Thông lượng mỗi chạy | ~kb | ~5M SNP | ~6 Tb (NovaSeq X) | ~30-60 Gb | ~200 Gb (PromethION) |
| Chi phí/Gb (2024) | ~$2000 | N/A | ~$3-5 | ~$25-30 | ~$15-20 |
| Phát hiện SV | Hạn chế | Không | Một phần | Xuất sắc | Xuất sắc |
| Methylation trực tiếp | Không | Không | Không (bisulfite) | Có (PacBio Revio) | Có |
| Di động | Không | Không | Hạn chế | Không | Có (MinION) |
| Nhược điểm chính | Thông lượng thấp | Chỉ biến thể đã biết | Không đọc lặp dài | Chi phí/Gb cao hơn | Lỗi ngẫu nhiên mỗi đọc |
Bảng 7.1. So sánh đặc tính kỹ thuật các nền tảng genomics chính (thông số tham chiếu năm 2024).
7.2. Xu Hướng Hội Tụ: Pangenome và Multimodal Sequencing
Không thể kết thúc bức tranh tổng quan mà không đề cập đến hai xu hướng đang định hình thập kỷ 2020: pangenome và giải trình tự đa phương thức.
Pangenome (pangenome: biểu diễn toàn bộ sự đa dạng di truyền của một loài dưới dạng đồ thị thay vì một trình tự đơn) là sự thừa nhận rằng mọi genome “tham chiếu” duy nhất đều thiên vị về mặt quần thể. Genome GRCh38 chủ yếu đến từ một số ít cá thể người Bắc Mỹ gốc Âu. Dự án Human Pangenome Reference Consortium đã trình bày phiên bản pangenome đầu tiên năm 2023, xây dựng từ genome của 47 cá thể đa dạng toàn cầu bằng công nghệ PacBio HiFi và ONT, lần đầu tiên biểu diễn sự đa dạng di truyền loài người như một đồ thị biếnthể (variation graph) thay vì một tham chiếu tuyến tính (Liao et al., 2023).
Giải trình tự đa phương thức (multimodal sequencing) là xu hướng kết hợp trong cùng một thí nghiệm nhiều loại thông tin trước đây cần quy trình riêng biệt. Ví dụ, long-read bisulfite-free methylation từ ONT cho phép đọc đồng thời trình tự nucleotide, trạng thái 5mC và 6mA, và cấu trúc haplotype từ một mẫu dữ liệu duy nhất. Single-cell long-read kết hợp phân giải tế bào đơn với độ dài đọc đủ để xác định toàn bộ isoform RNA-seq mà không cần reconstruction tính toán.
Kết Luận
Hành trình từ Sanger đến long-read không phải là một đường thẳng cải tiến đơn thuần mà là một chuỗi các cuộc cách mạng mô hình tư duy (paradigm shift): từ đọc để biết trình tự, sang so sánh để biết sự khác biệt quần thể, sang giải trình tự đại trà để biết toàn bộ biến thể cá thể, và cuối cùng sang đọc dài để hiểu cấu trúc, haplotype và epigenetics trong bối cảnh toàn bộ nhiễm sắc thể.
Không công nghệ nào làm obsolete (lỗi thời) hoàn toàn công nghệ trước đó. Sanger vẫn là tiêu chuẩn vàng cho xác nhận biến thể đơn lẻ độ chính xác cao. SNP array vẫn là lựa chọn kinh tế nhất cho GWAS quy mô lớn với hàng trăm nghìn mẫu. Illumina NGS vẫn là nền tảng mặc định cho RNA-seq và ứng dụng cần độ sâu đọc cao với chi phí thấp. Long-read ngày càng chiếm ưu thế trong các ứng dụng đòi hỏi lắp ghép hoàn chỉnh, phát hiện structural variant, và epigenomics trực tiếp.
Điểm hội tụ của tất cả các công nghệ vẫn là reference genome như hệ toạ độ dùng chung và xu hướng hiện tại hướng đến pangenome như cách biểu diễn toàn diện hơn sự đa dạng sinh học của loài người. Bài tutorial đi kèm sẽ hướng dẫn cách làm việc thực hành với dữ liệu từ mỗi loại công nghệ kể trên.
Tài Liệu Tham Khảo
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., Hall, K. P., Evers, D. J., Barnes, C. L., Bignell, H. R., Boutell, J. M., Bryant, J., Carter, R. J., Keira Cheetham, R., Cox, A. J., Ellis, D. J., Flatbush, M. R., Gormley, N. A., Humphray, S. J., … Smith, A. J. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature, 456(7218), 53–59. https://doi.org/10.1038/nature07517
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., Peluso, P., Rank, D., Baybayan, P., Bettman, B., Bibillo, A., Bjornson, K., Chaudhuri, B., Christians, F., Cicero, R., Clark, S., Dalal, R., Dewinter, A., Dixon, J., … Turner, S. (2009). Real-time DNA sequencing from single polymerase molecules. Science, 323(5910), 133–138. https://doi.org/10.1126/science.1162986
International Human Genome Sequencing Consortium. (2004). Finishing the euchromatic sequence of the human genome. Nature, 431(7011), 931–945. https://doi.org/10.1038/nature03001
Liao, W. W., Asri, M., Ebler, J., Doerr, D., Haukness, M., Hickey, G., Lu, S., Lucas, J. K., Monlong, J., Abel, H. J., Buonaiuto, S., Chang, X. H., Cheng, H., Chu, J., Colonna, V., Ebert, P., Feng, X., Fischer, C., Fulton, R. S., … Paten, B. (2023). A draft human pangenome reference. Nature, 617(7960), 312–324. https://doi.org/10.1038/s41586-023-05896-x
Loman, N. J., Quick, J., & Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nature Methods, 12(8), 733–735. https://doi.org/10.1038/nmeth.3444
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., Berka, J., Braverman, M. S., Chen, Y. J., Chen, Z., Dewell, S. B., Du, L., Fierro, J. M., Gomes, X. V., Godwin, B. C., He, W., Helgesen, S., Ho, C. H., Irzyk, G. P., … Rothberg, J. M. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature, 437(7057), 376–380. https://doi.org/10.1038/nature03959
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., Vollger, M. R., Altemose, N., Uralsky, L., Gershman, A., Aganezov, S., Hoyt, S. J., Diekhans, M., Logsdon, G. A., Alonge, M., Antonarakis, S. E., Borchers, M., Bouffard, G. G., Brooks, S. Y., … Phillippy, A. M. (2022). The complete sequence of a human genome. Science, 376(6588), 44–53. https://doi.org/10.1126/science.abj6987
Sanger, F., Nicklen, S., & Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences, 74(12), 5463–5467. https://doi.org/10.1073/pnas.74.12.5463
Schena, M., Shalon, D., Davis, R. W., & Brown, P. O. (1995). Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science, 270(5235), 467–470. https://doi.org/10.1126/science.270.5235.467
Wellcome Trust Case Control Consortium. (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 447(7145), 661–678. https://doi.org/10.1038/nature05911
Explore more like this
Gene Regulatory Network: Từ Phân Tử Đến Multi-Omics Và Ứng Dụng Lâm Sàng
Genome của người chứa khoảng 20.000 gene mã hóa protein, nhưng không phải gene nào cũng hoạt động ở mọi tế bào trong mọi thời điểm. Một tế bào gan,...
Từ Dữ Liệu Thô Đến Biến Thể Di Truyền: Genome Alignment, Variant Calling, QC và Annotation
Khi một mẫu DNA được đưa vào máy giải trình tự, kết quả trả về không phải là một câu trả lời hoàn chỉnh về bộ genome của cá thể...
Transcriptomics: Hành Trình Giải Mã Hệ Phiên Mã và Các Công Nghệ Chủ Chốt
Tế bào người chứa khoảng 3,2 tỷ cặp bazơ DNA — nhưng không phải tất cả đều được “đọc” cùng một lúc, hay ở mọi loại tế bào. Một tế...
Comments